

Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease
- PMID: 32251296
- PMCID: PMC7089947
- DOI: 10.1038/s41467-020-15342-5
Colonic microbiota is associated with inflammation and host epigenomic alterations in inflammatory bowel disease
Abstract
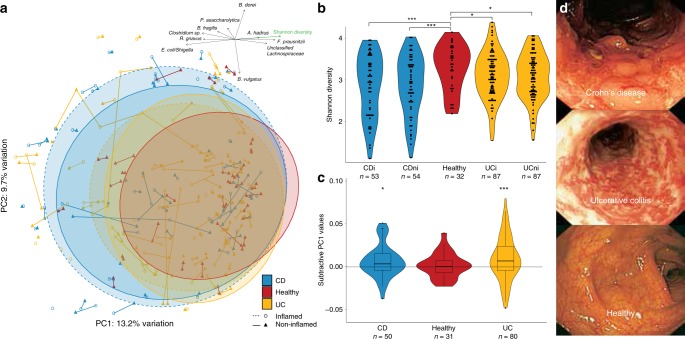
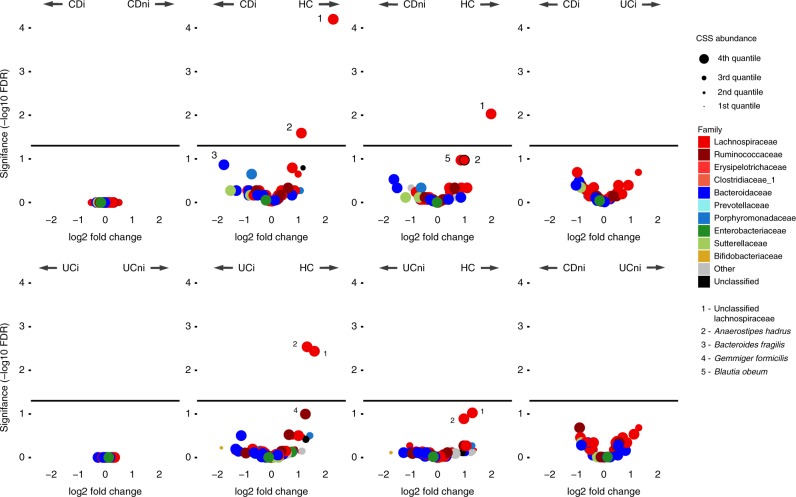
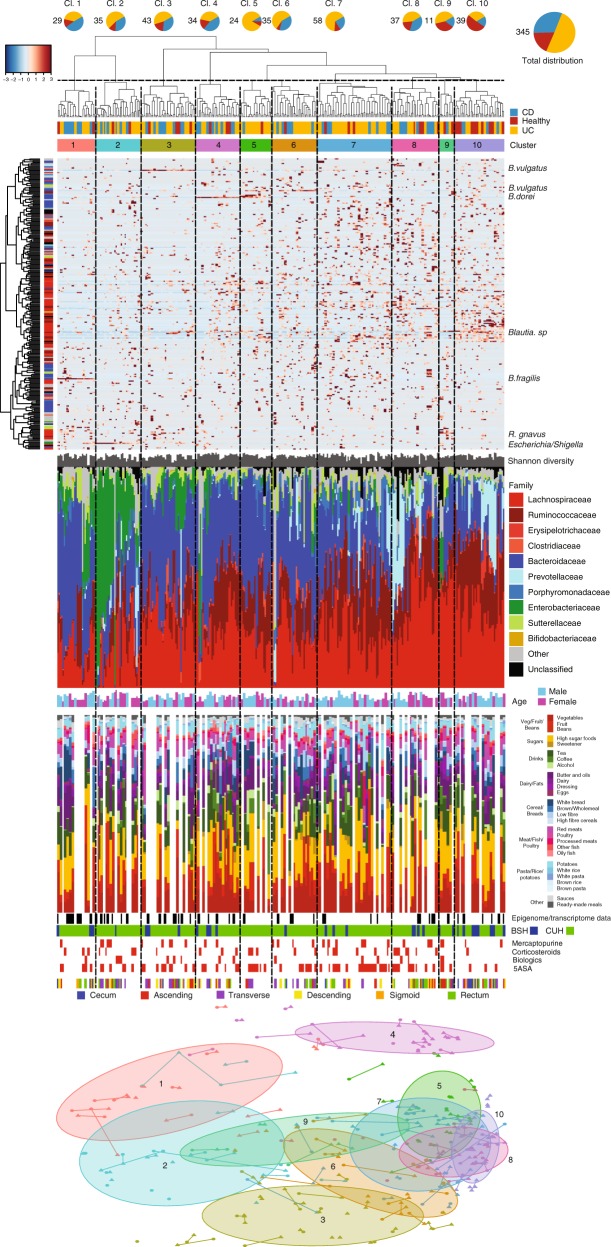
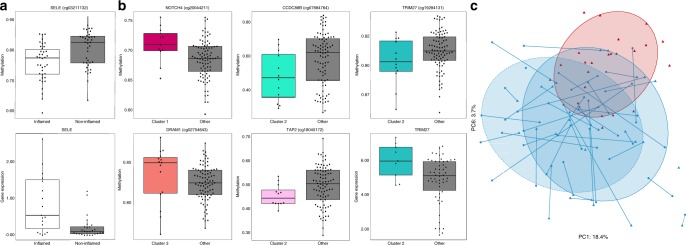
Studies of inflammatory bowel disease (IBD) have been inconclusive in relating microbiota with distribution of inflammation. We report microbiota, host transcriptomics, epigenomics and genetics from matched inflamed and non-inflamed colonic mucosa [50 Crohn's disease (CD); 80 ulcerative colitis (UC); 31 controls]. Changes in community-wide and within-patient microbiota are linked with inflammation, but we find no evidence for a distinct microbial diagnostic signature, probably due to heterogeneous host-microbe interactions, and show only marginal microbiota associations with habitual diet. Epithelial DNA methylation improves disease classification and is associated with both inflammation and microbiota composition. Microbiota sub-groups are driven by dominant Enterbacteriaceae and Bacteroides species, representative strains of which are pro-inflammatory in vitro, are also associated with immune-related epigenetic markers. In conclusion, inflamed and non-inflamed colonic segments in both CD and UC differ in microbiota composition and epigenetic profiles.
Conflict of interest statement
This work was supported in part by Second Genome; T.Z.D. is a co-founder and Vice-president of Second Genome; P.J.M., S.I., and C.C.A. were employees of Second Genome at the time of the analysis.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
