

Structure and function of polycystin channels in primary cilia
- PMID: 32251715
- PMCID: PMC7373203
- DOI: 10.1016/j.cellsig.2020.109626
Structure and function of polycystin channels in primary cilia
Abstract
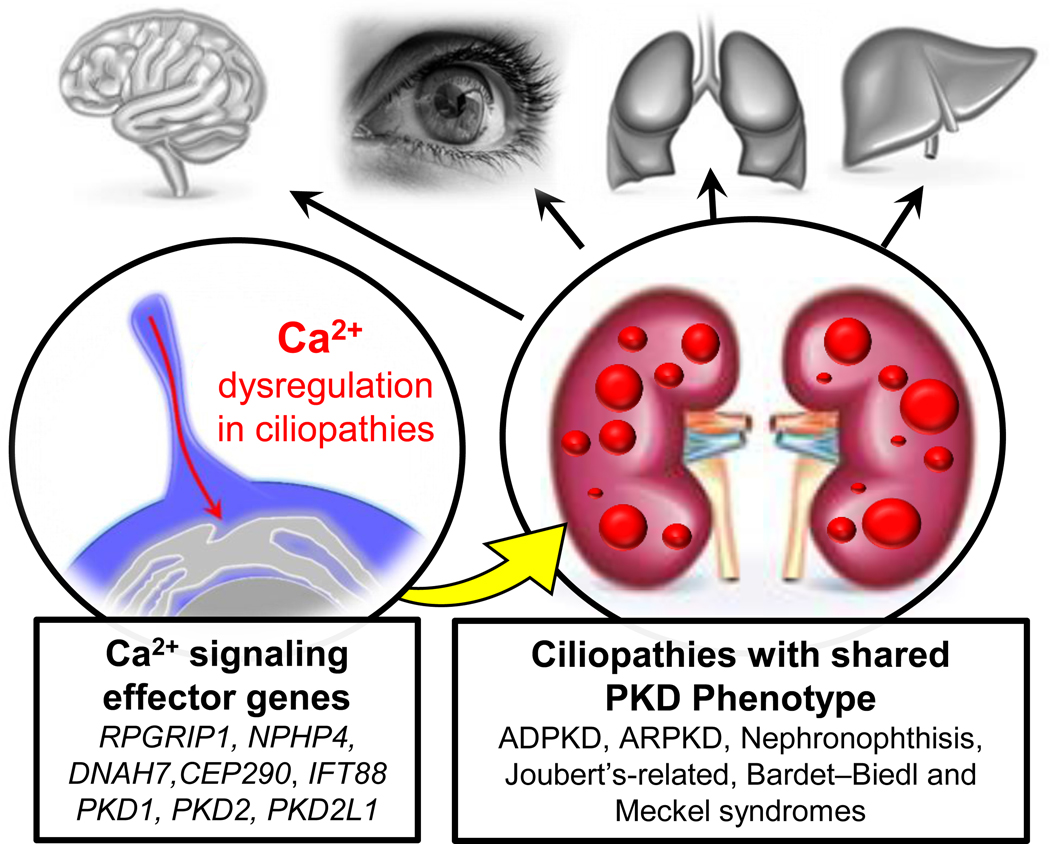
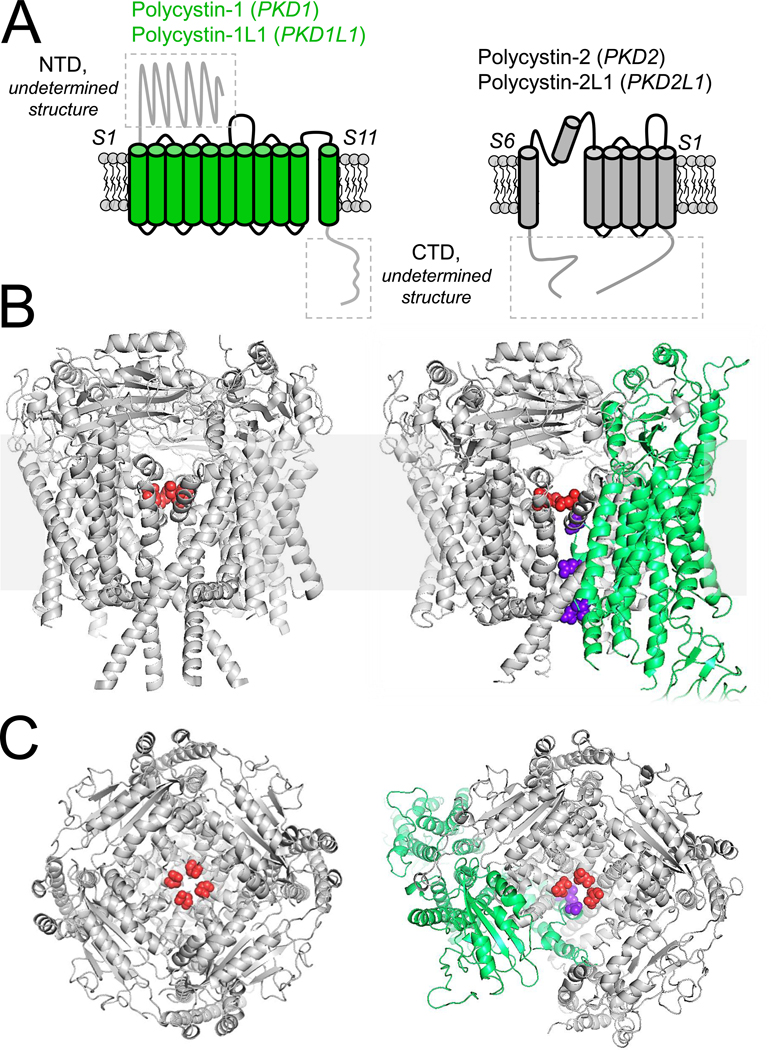
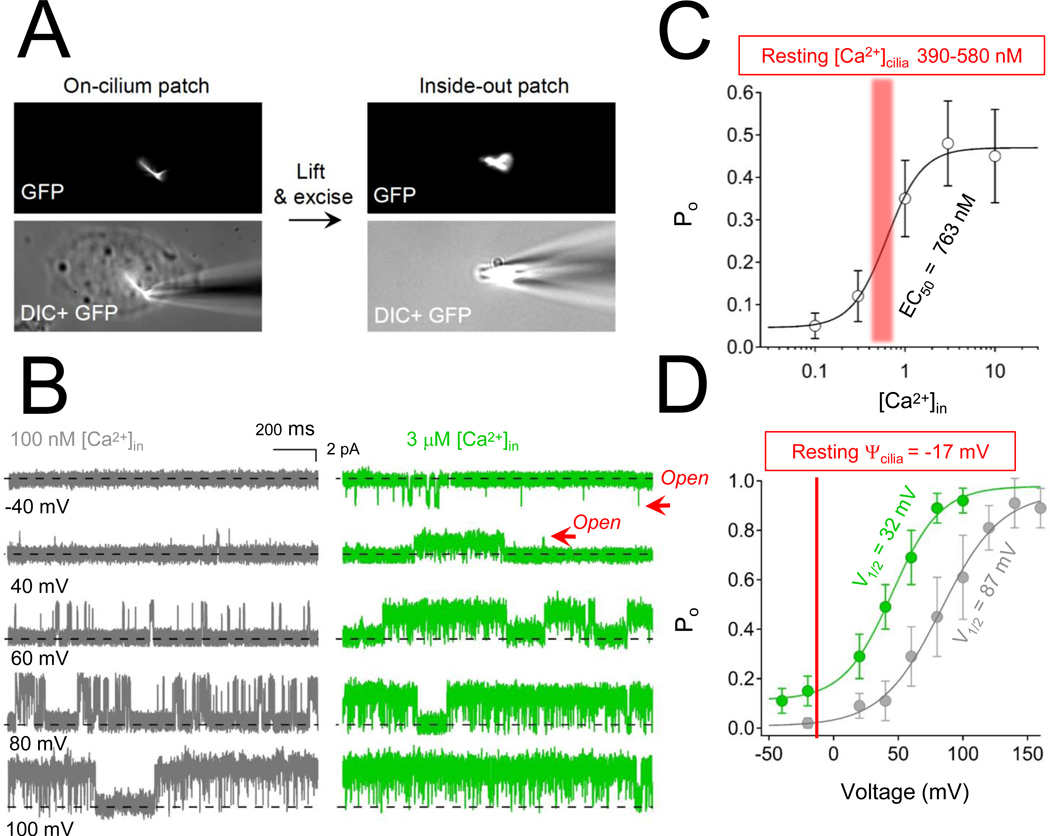
Variants in genes which encode for polycystin-1 and polycystin-2 cause most forms of autosomal dominant polycystic disease (ADPKD). Despite our strong understanding of the genetic determinants of ADPKD, we do not understand the structural features which govern the function of polycystins at the molecular level, nor do we understand the impact of most disease-causing variants on the conformational state of these proteins. These questions have remained elusive because polycystins localize to several organelle membranes, including the primary cilia. Primary cilia are microtubule based organelles which function as cellular antennae. Polycystin-2 and related polycystin-2 L1 are members of the transient receptor potential (TRP) ion channel family, and form distinct ion channels in the primary cilia of disparate cell types which can be directly measured. Polycystin-1 has both ion channel and adhesion G-protein coupled receptor (GPCR) features-but its role in forming a channel complex or as a channel subunit chaperone is undetermined. Nonetheless, recent polycystin structural determination by cryo-EM has provided a molecular template to understand their biophysical regulation and the impact of disease-causing variants. We will review these advances and discuss hypotheses regarding the regulation of polycystin channel opening by their structural domains within the context of the primary cilia.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Mochizuki T et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272, 1339–1342 (1996). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
