

Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins
- PMID: 32252787
- PMCID: PMC7137325
- DOI: 10.1186/s13059-020-01982-9
Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins
Abstract
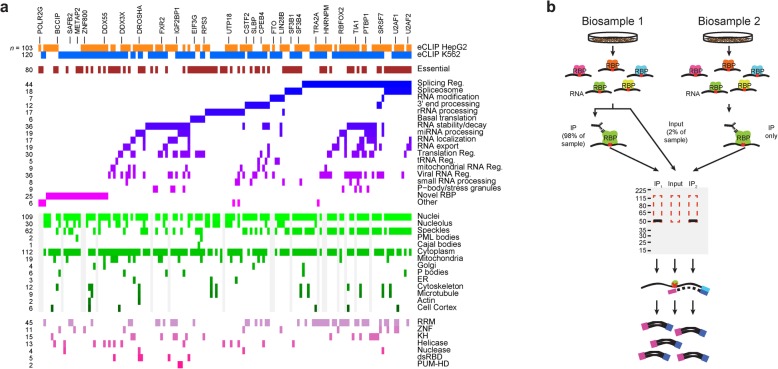
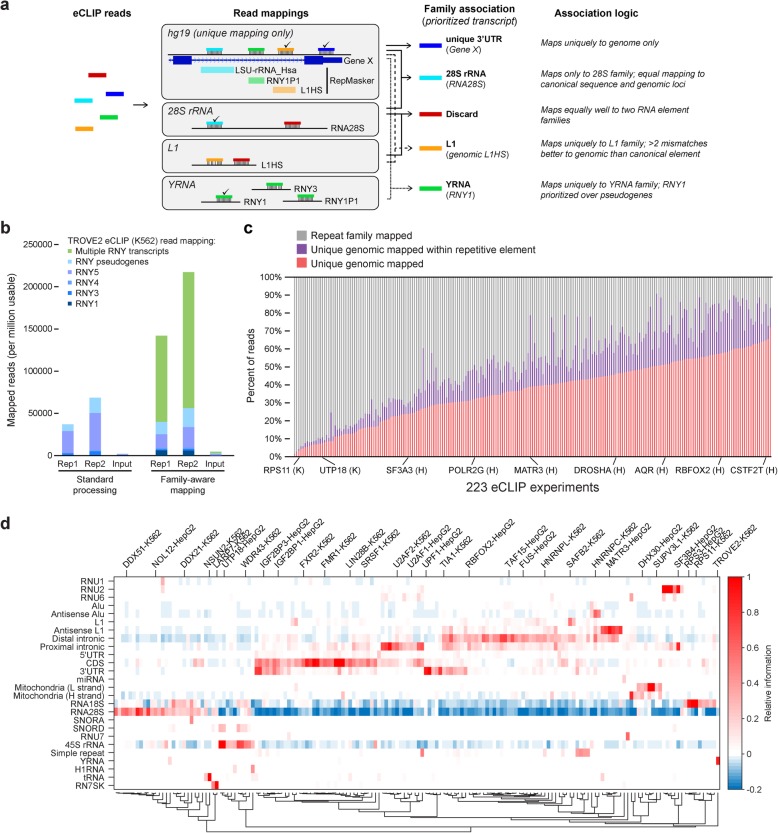
Background: A critical step in uncovering rules of RNA processing is to study the in vivo regulatory networks of RNA binding proteins (RBPs). Crosslinking and immunoprecipitation (CLIP) methods enable mapping RBP targets transcriptome-wide, but methodological differences present challenges to large-scale analysis across datasets. The development of enhanced CLIP (eCLIP) enabled the mapping of targets for 150 RBPs in K562 and HepG2, creating a unique resource of RBP interactomes profiled with a standardized methodology in the same cell types.
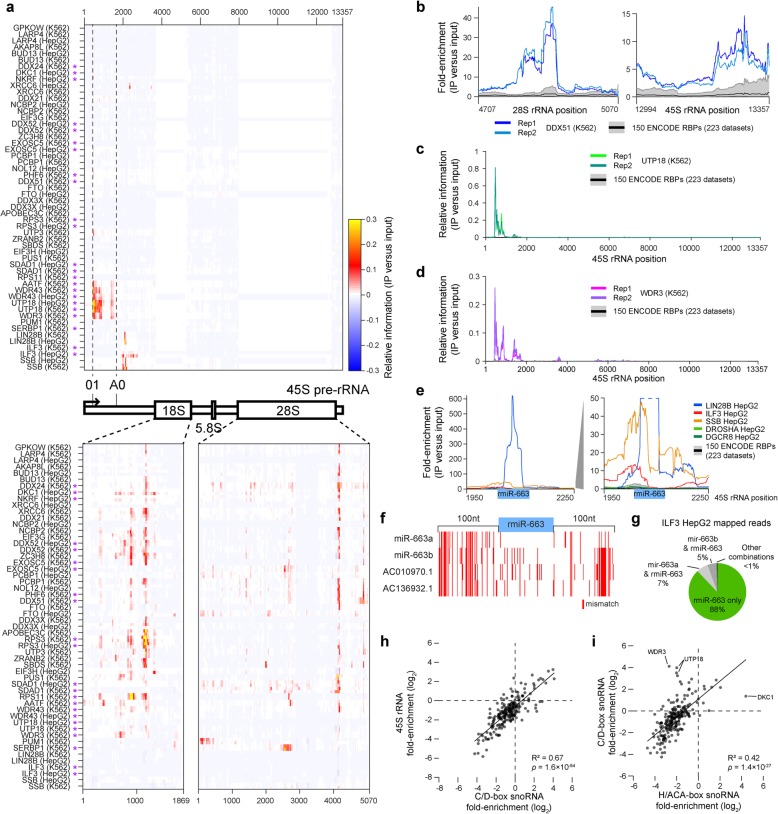
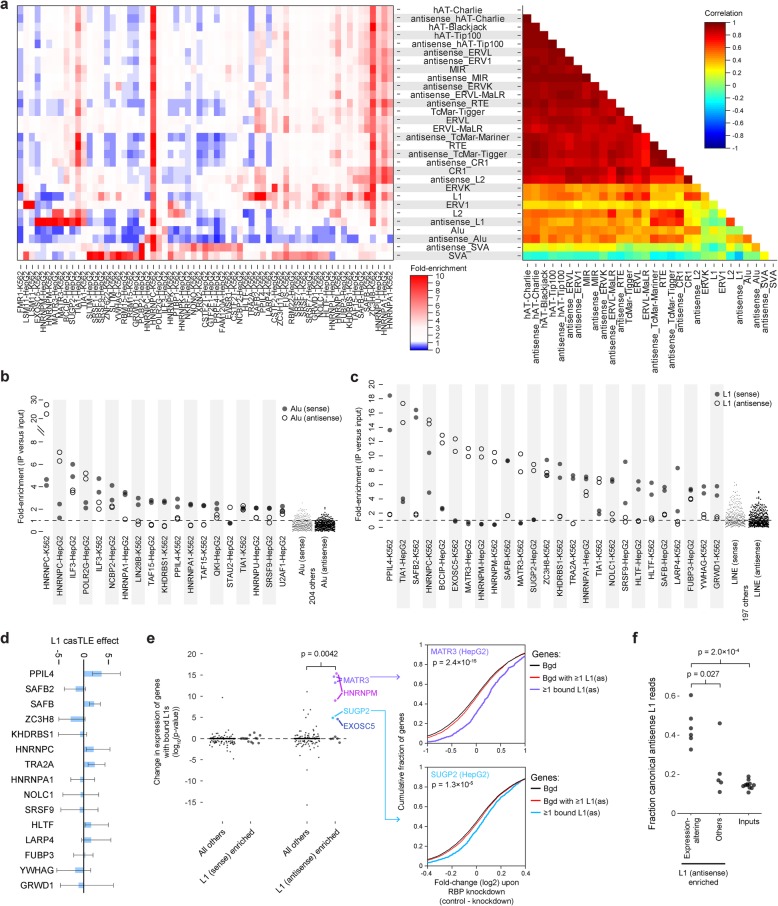
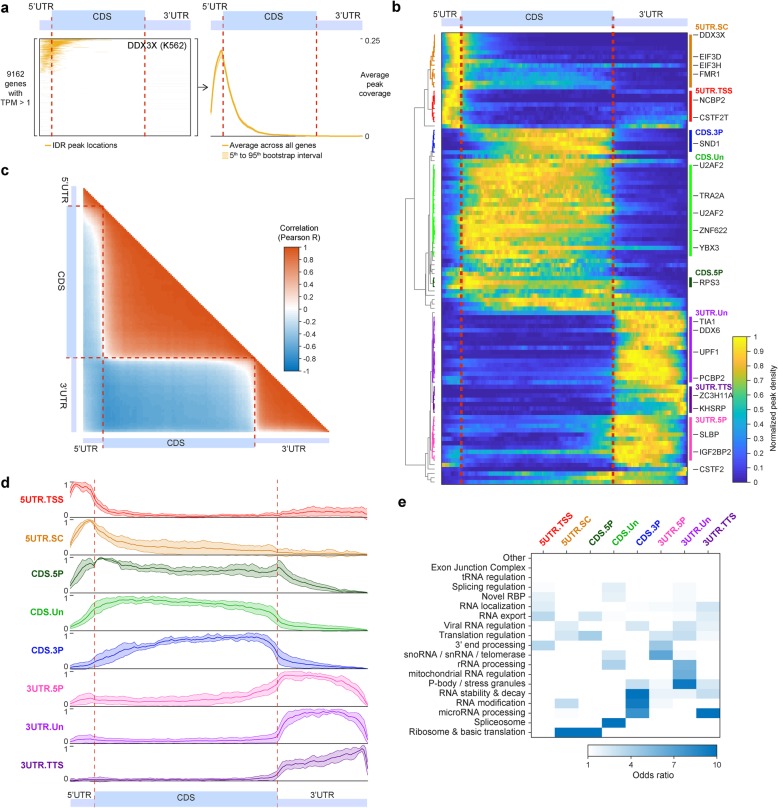
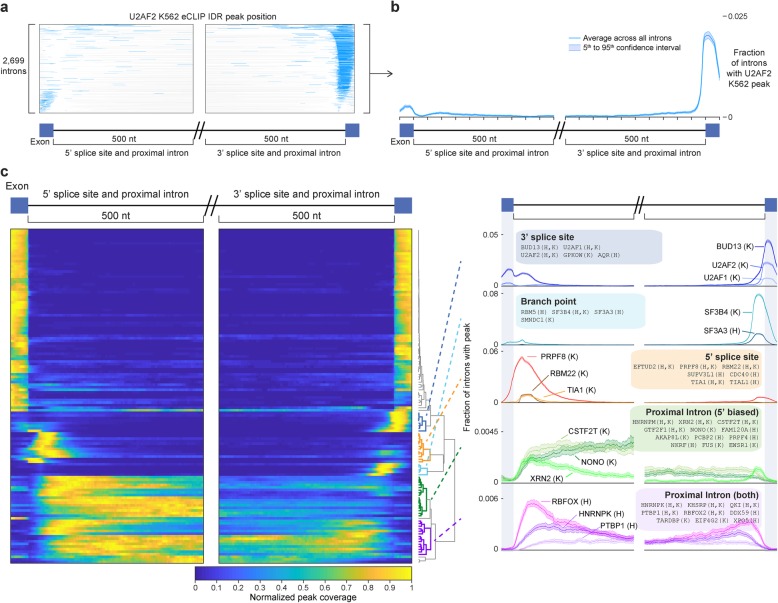
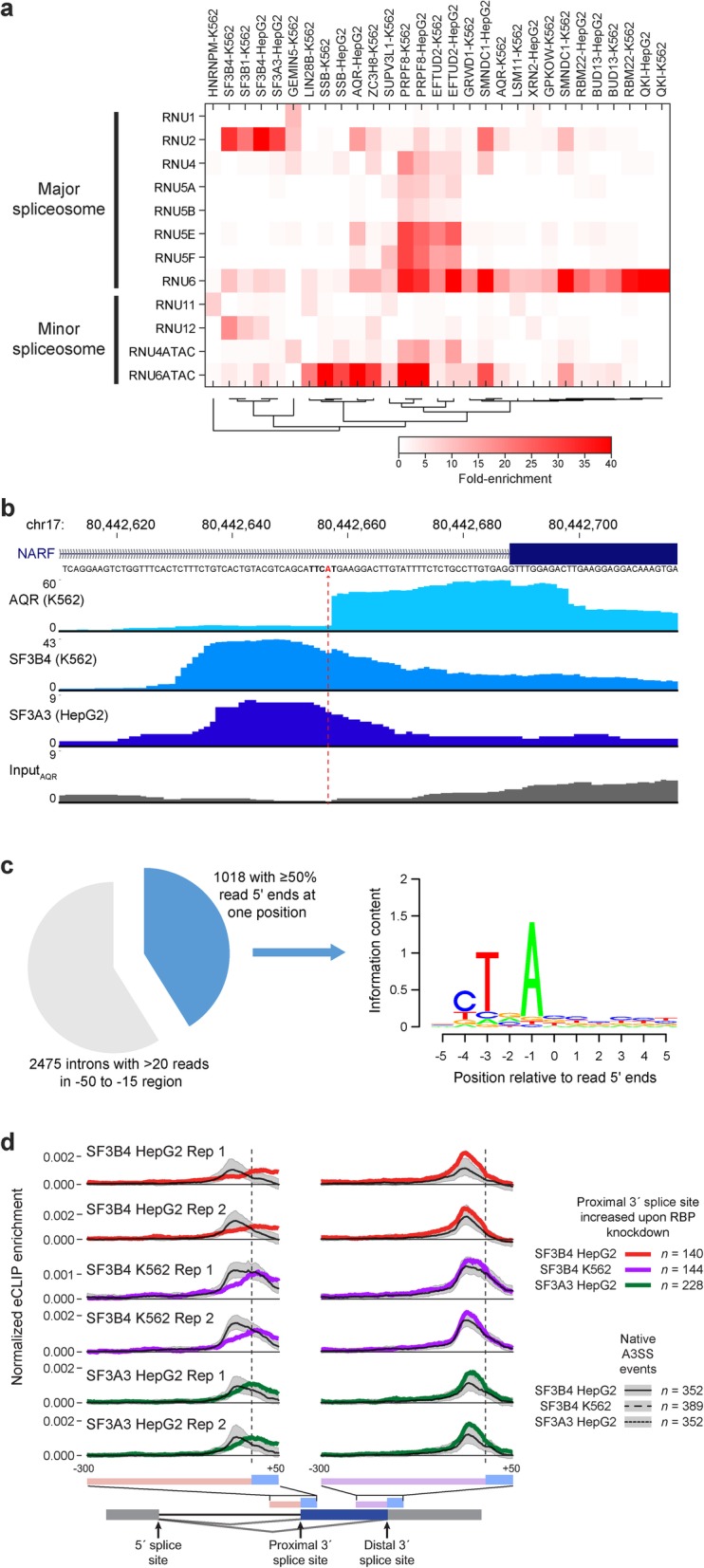
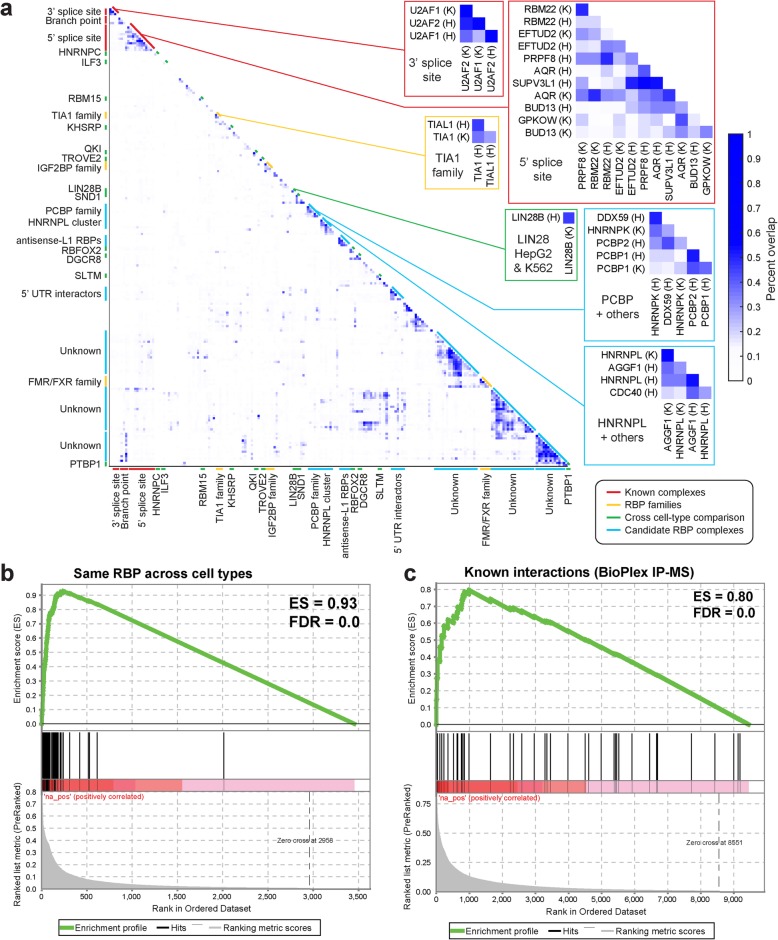
Results: Our analysis of 223 eCLIP datasets reveals a range of binding modalities, including highly resolved positioning around splicing signals and mRNA untranslated regions that associate with distinct RBP functions. Quantification of enrichment for repetitive and abundant multicopy elements reveals 70% of RBPs have enrichment for non-mRNA element classes, enables identification of novel ribosomal RNA processing factors and sites, and suggests that association with retrotransposable elements reflects multiple RBP mechanisms of action. Analysis of spliceosomal RBPs indicates that eCLIP resolves AQR association after intronic lariat formation, enabling identification of branch points with single-nucleotide resolution, and provides genome-wide validation for a branch point-based scanning model for 3' splice site recognition. Finally, we show that eCLIP peak co-occurrences across RBPs enable the discovery of novel co-interacting RBPs.
Conclusions: This work reveals novel insights into RNA biology by integrated analysis of eCLIP profiling of 150 RBPs with distinct functions. Further, our quantification of both mRNA and other element association will enable further research to identify novel roles of RBPs in regulating RNA processing.
Keywords: CLIP-seq; RNA binding protein; RNA processing; eCLIP.
Conflict of interest statement
ELVN is co-founder, member of the Board of Directors, on the SAB, equity holder, and paid consultant for Eclipse BioInnovations. GWY is co-founder, member of the Board of Directors, on the SAB, equity holder, and paid consultant for Locana and Eclipse BioInnovations. GWY is a visiting professor at the National University of Singapore. ELVN's and GWY's interests have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. The other authors declare that they have no competing interests.
Figures
References
-
- Martinez FJ, Pratt GA, Van Nostrand EL, Batra R, Huelga SC, Kapeli K, Freese P, Chun SJ, Ling K, Gelboin-Burkhart C, et al. Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron. 2016;92:780–795. doi: 10.1016/j.neuron.2016.09.050. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
