

Bayesian integrative analysis of epigenomic and transcriptomic data identifies Alzheimer's disease candidate genes and networks
- PMID: 32255787
- PMCID: PMC7138305
- DOI: 10.1371/journal.pcbi.1007771
Bayesian integrative analysis of epigenomic and transcriptomic data identifies Alzheimer's disease candidate genes and networks
Abstract
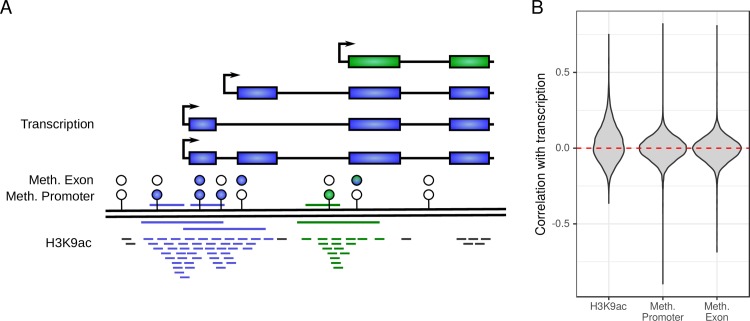
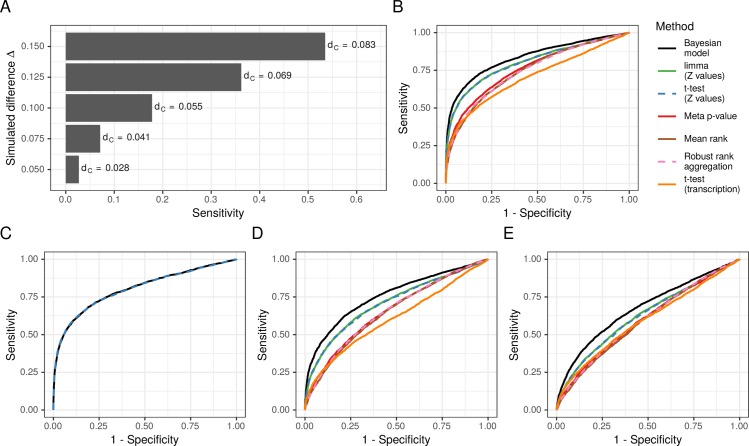
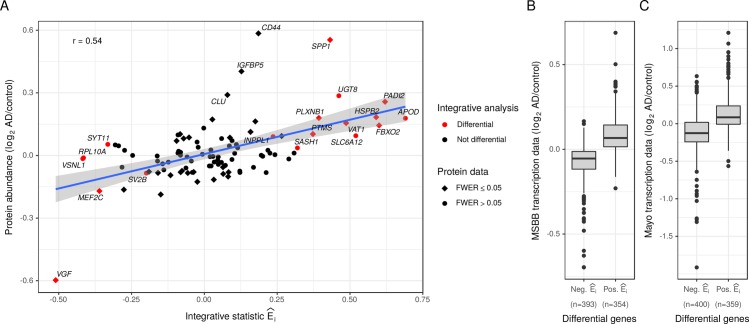
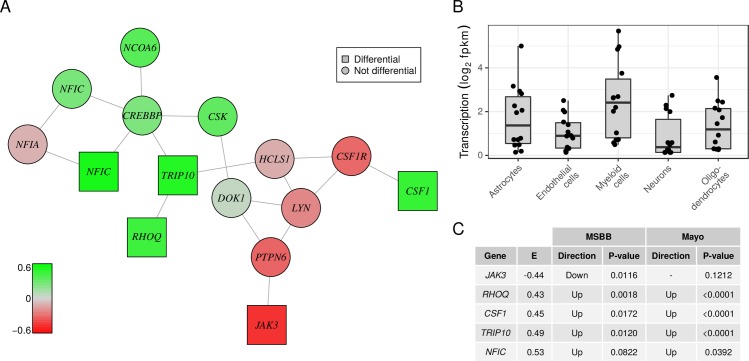
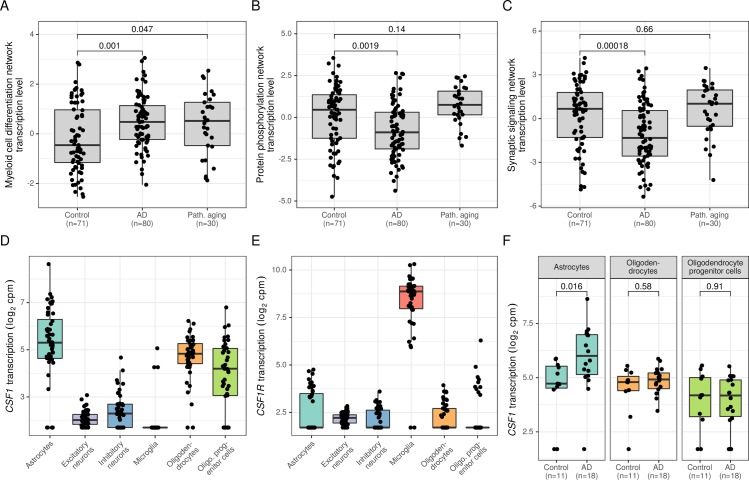
Biomedical research studies have generated large multi-omic datasets to study complex diseases like Alzheimer's disease (AD). An important aim of these studies is the identification of candidate genes that demonstrate congruent disease-related alterations across the different data types measured by the study. We developed a new method to detect such candidate genes in large multi-omic case-control studies that measure multiple data types in the same set of samples. The method is based on a gene-centric integrative coefficient quantifying to what degree consistent differences are observed in the different data types. For statistical inference, a Bayesian hierarchical model is used to study the distribution of the integrative coefficient. The model employs a conditional autoregressive prior to integrate a functional gene network and to share information between genes known to be functionally related. We applied the method to an AD dataset consisting of histone acetylation, DNA methylation, and RNA transcription data from human cortical tissue samples of 233 subjects, and we detected 816 genes with consistent differences between persons with AD and controls. The findings were validated in protein data and in RNA transcription data from two independent AD studies. Finally, we found three subnetworks of jointly dysregulated genes within the functional gene network which capture three distinct biological processes: myeloid cell differentiation, protein phosphorylation and synaptic signaling. Further investigation of the myeloid network indicated an upregulation of this network in early stages of AD prior to accumulation of hyperphosphorylated tau and suggested that increased CSF1 transcription in astrocytes may contribute to microglial activation in AD. Thus, we developed a method that integrates multiple data types and external knowledge of gene function to detect candidate genes, applied the method to an AD dataset, and identified several disease-related genes and processes demonstrating the usefulness of the integrative approach.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
