

Molecular and Cellular Pathways Contributing to Joint Damage in Rheumatoid Arthritis
- PMID: 32256192
- PMCID: PMC7103059
- DOI: 10.1155/2020/3830212
Molecular and Cellular Pathways Contributing to Joint Damage in Rheumatoid Arthritis
Abstract
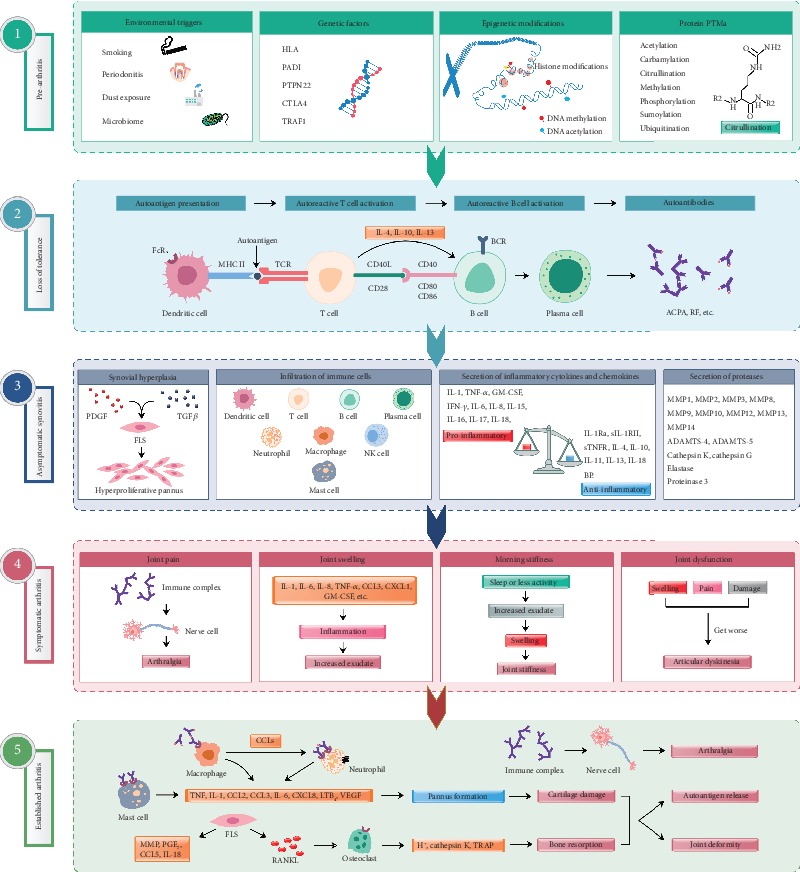
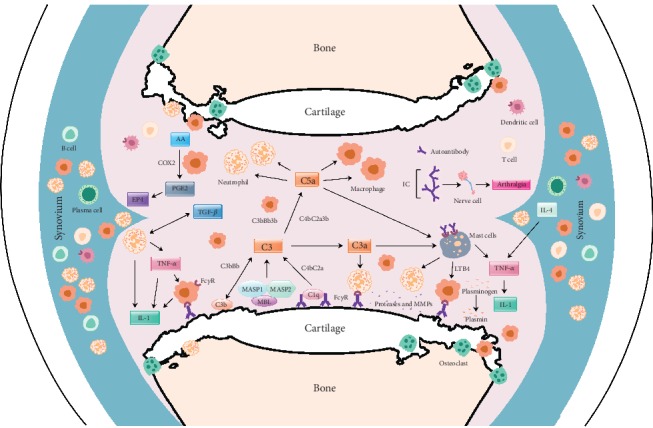
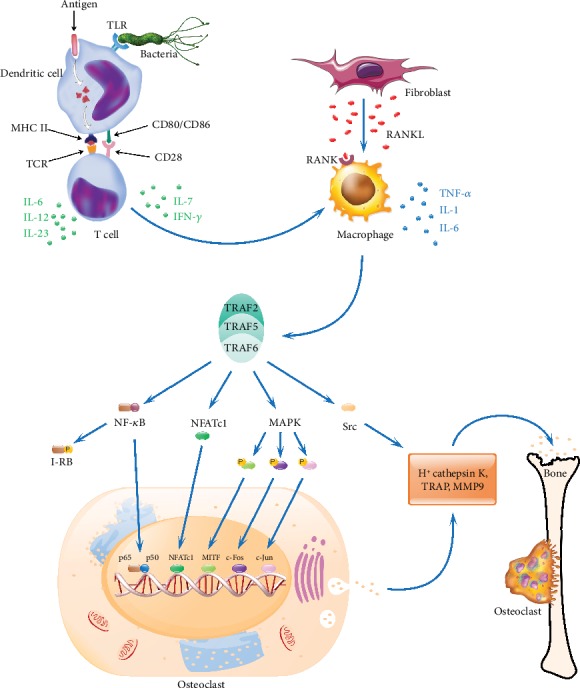
Rheumatoid arthritis is a chronic autoimmune syndrome associated with several genetic, epigenetic, and environmental factors affecting the articular joints contributing to cartilage and bone damage. Although etiology of this disease is not clear, several immune pathways, involving immune (T cells, B cells, dendritic cells, macrophages, and neutrophils) and nonimmune (fibroblasts and chondrocytes) cells, participate in the secretion of many proinflammatory cytokines, chemokines, proteases (MMPs, ADAMTS), and other matrix lysing enzymes that could disturb the immune balance leading to cartilage and bone damage. The presence of autoantibodies preceding the clinical onset of arthritis and the induction of bone erosion early in the disease course clearly suggest that initiation events damaging the cartilage and bone start very early during the autoimmune phase of the arthritis development. During this process, several signaling molecules (RANKL-RANK, NF-κB, MAPK, NFATc1, and Src kinase) are activated in the osteoclasts, cells responsible for bone resorption. Hence, comprehensive knowledge on pathogenesis is a prerequisite for prevention and development of targeted clinical treatment for RA patients that can restore the immune balance improving clinical therapy.
Copyright © 2020 Qinghua Fang et al.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
