

Inherited iron overload disorders
- PMID: 32258529
- PMCID: PMC7063521
- DOI: 10.21037/tgh.2019.11.15
Inherited iron overload disorders
Abstract
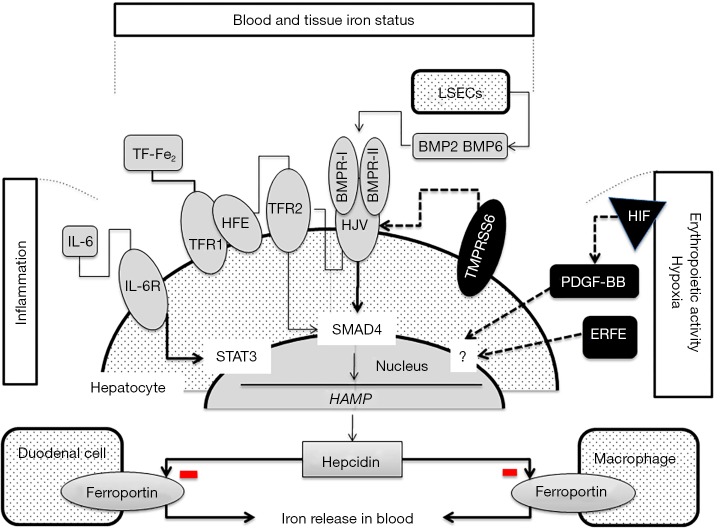
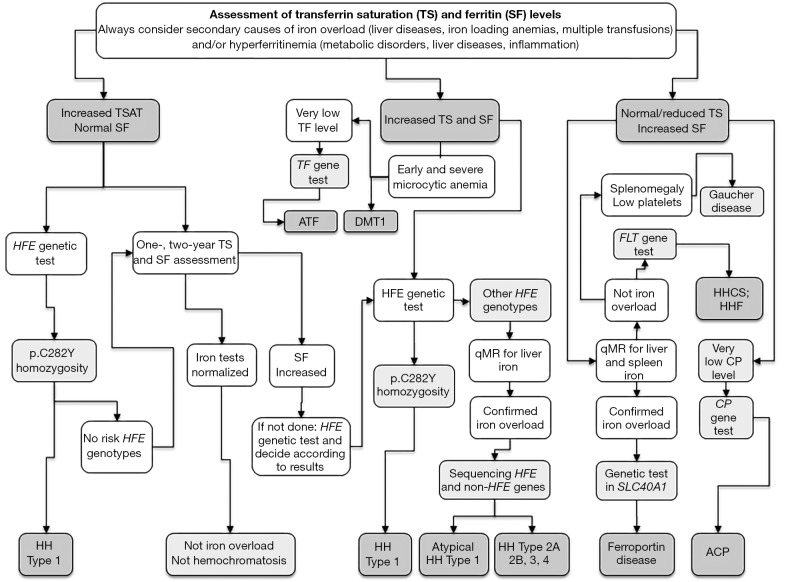
Hereditary iron overload includes several disorders characterized by iron accumulation in tissues, organs, or even single cells or subcellular compartments. They are determined by mutations in genes directly involved in hepcidin regulation, cellular iron uptake, management and export, iron transport and storage. Systemic forms are characterized by increased serum ferritin with or without high transferrin saturation, and with or without functional iron deficient anemia. Hemochromatosis includes five different genetic forms all characterized by high transferrin saturation and serum ferritin, but with different penetrance and expression. Mutations in HFE, HFE2, HAMP and TFR2 lead to inadequate or severely reduced hepcidin synthesis that, in turn, induces increased intestinal iron absorption and macrophage iron release leading to tissue iron overload. The severity of hepcidin down-regulation defines the severity of iron overload and clinical complications. Hemochromatosis type 4 is caused by dominant gain-of-function mutations of ferroportin preventing hepcidin-ferroportin binding and leading to hepcidin resistance. Ferroportin disease is due to loss-of-function mutation of SLC40A1 that impairs the iron export efficiency of ferroportin, causes iron retention in reticuloendothelial cell and hyperferritinemia with normal transferrin saturation. Aceruloplasminemia is caused by defective iron release from storage and lead to mild microcytic anemia, low serum iron, and iron retention in several organs including the brain, causing severe neurological manifestations. Atransferrinemia and DMT1 deficiency are characterized by iron deficient erythropoiesis, severe microcytic anemia with high transferrin saturation and parenchymal iron overload due to secondary hepcidin suppression. Diagnosis of the different forms of hereditary iron overload disorders involves a sequential strategy that combines clinical, imaging, biochemical, and genetic data. Management of iron overload relies on two main therapies: blood removal and iron chelators. Specific therapeutic options are indicated in patients with atransferrinemia, DMT1 deficiency and aceruloplasminemia.
Keywords: Iron overload; ferritin; transferrin saturation.
2020 Translational Gastroenterology and Hepatology. All rights reserved.
Conflict of interest statement
Conflicts of Interest: The authors have no conflicts of interest to declare.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources