

Pathogenesis of Nonalcoholic Steatohepatitis: An Overview
- PMID: 32258944
- PMCID: PMC7109346
- DOI: 10.1002/hep4.1479
Pathogenesis of Nonalcoholic Steatohepatitis: An Overview
Abstract
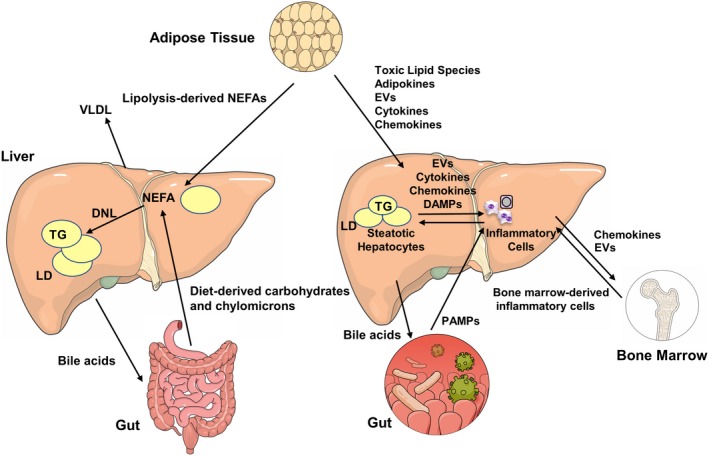
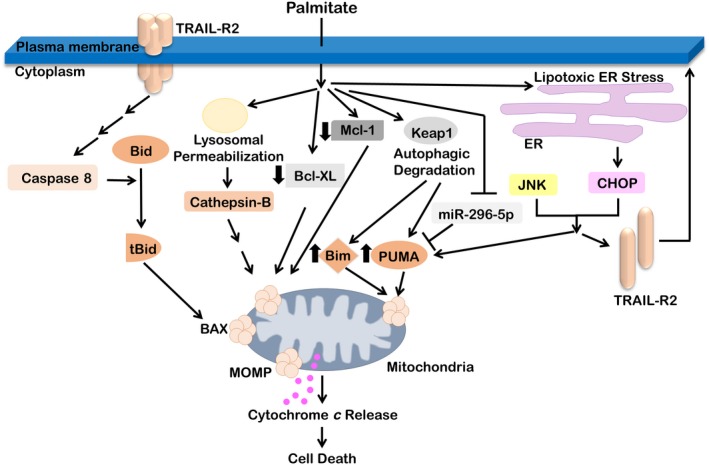
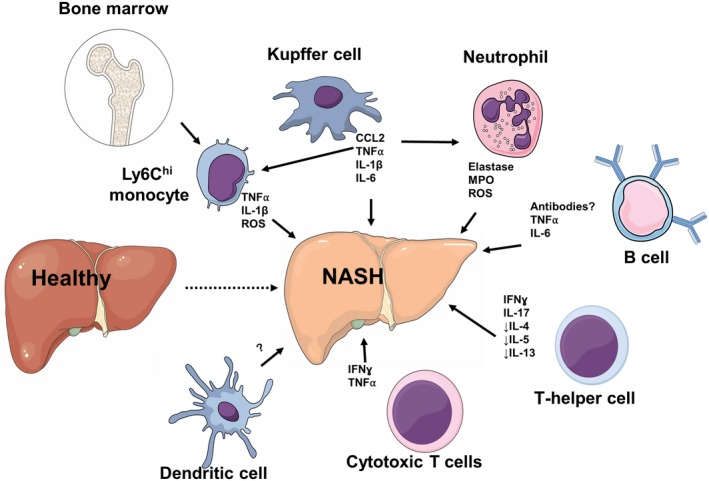
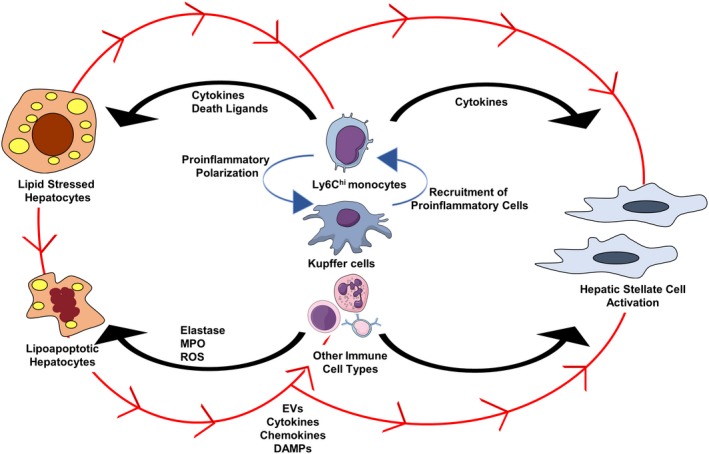
Nonalcoholic fatty liver disease (NAFLD) is a heterogeneous group of liver diseases characterized by the accumulation of fat in the liver. The heterogeneity of NAFLD is reflected in a clinical and histologic spectrum where some patients develop isolated steatosis of the liver, termed nonalcoholic fatty liver, whereas others develop hepatocyte injury, ballooning, inflammation, and consequent fibrosis, termed nonalcoholic steatohepatitis (NASH). Systemic insulin resistance is a major driver of hepatic steatosis in NAFLD. Lipotoxicity of accumulated lipids along with activation of the innate immune system are major drivers of NASH. Lipid-induced sublethal and lethal stress culminates in the activation of inflammatory processes, such as the release of proinflammatory extracellular vesicles and cell death. Innate and adaptive immune mechanisms involving macrophages, dendritic cells, and lymphocytes are central drivers of inflammation that recognize damage- and pathogen-associated molecular patterns and contribute to the progression of the inflammatory cascade. While the activation of the innate immune system and the recruitment of proinflammatory monocytes into the liver in NASH are well known, the exact signals that lead to this remain less well defined. Further, the contribution of other immune cell types, such as neutrophils and B cells, is an area of intense research. Many host factors, such as the microbiome and gut-liver axis, modify individual susceptibility to NASH. In this review, we discuss lipotoxicity, inflammation, and the contribution of interorgan crosstalk in NASH pathogenesis.
© 2020 The Authors. Hepatology Communications published by Wiley Periodicals, Inc., on behalf of the American Association for the Study of Liver Diseases.
Figures
References
-
- Younossi ZM, Blissett D, Blissett R, Henry L, Stepanova M, Younossi Y, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016;64:1577‐1586. - PubMed
-
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. - PubMed
-
- Hagstrom H, Nasr P, Ekstedt M, Hammar U, Stal P, Hultcrantz R, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy‐proven NAFLD. J Hepatol 2017;67:1265‐1273. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
