

A multidimensional systems biology analysis of cellular senescence in aging and disease
- PMID: 32264951
- PMCID: PMC7333371
- DOI: 10.1186/s13059-020-01990-9
A multidimensional systems biology analysis of cellular senescence in aging and disease
Abstract
Background: Cellular senescence, a permanent state of replicative arrest in otherwise proliferating cells, is a hallmark of aging and has been linked to aging-related diseases. Many genes play a role in cellular senescence, yet a comprehensive understanding of its pathways is still lacking.
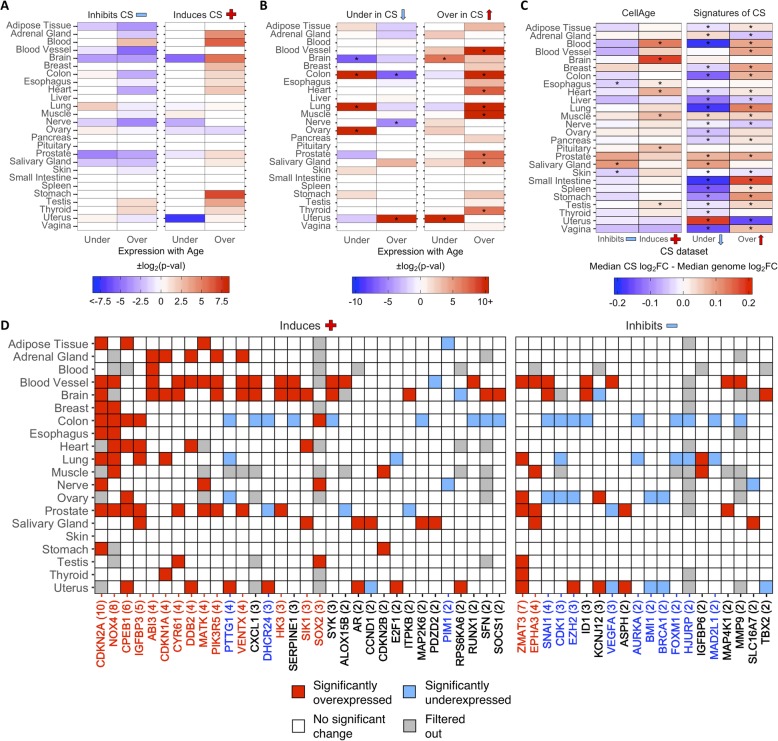
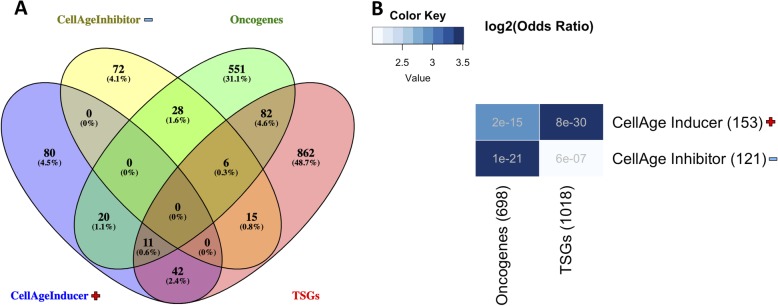
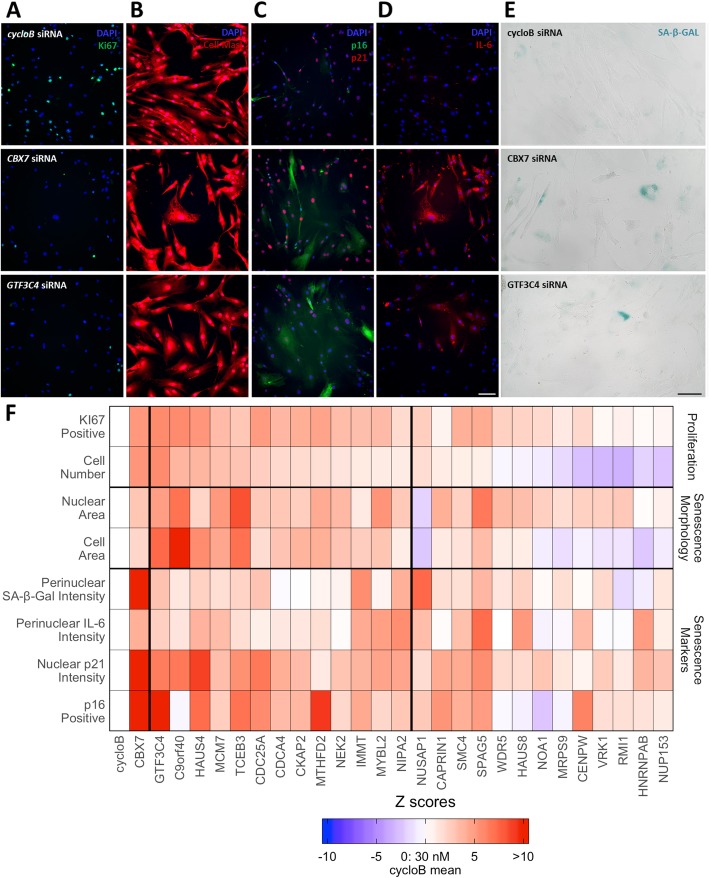
Results: We develop CellAge (http://genomics.senescence.info/cells), a manually curated database of 279 human genes driving cellular senescence, and perform various integrative analyses. Genes inducing cellular senescence tend to be overexpressed with age in human tissues and are significantly overrepresented in anti-longevity and tumor-suppressor genes, while genes inhibiting cellular senescence overlap with pro-longevity and oncogenes. Furthermore, cellular senescence genes are strongly conserved in mammals but not in invertebrates. We also build cellular senescence protein-protein interaction and co-expression networks. Clusters in the networks are enriched for cell cycle and immunological processes. Network topological parameters also reveal novel potential cellular senescence regulators. Using siRNAs, we observe that all 26 candidates tested induce at least one marker of senescence with 13 genes (C9orf40, CDC25A, CDCA4, CKAP2, GTF3C4, HAUS4, IMMT, MCM7, MTHFD2, MYBL2, NEK2, NIPA2, and TCEB3) decreasing cell number, activating p16/p21, and undergoing morphological changes that resemble cellular senescence.
Conclusions: Overall, our work provides a benchmark resource for researchers to study cellular senescence, and our systems biology analyses reveal new insights and gene regulators of cellular senescence.
Keywords: Biogerontology; Cancer; Genetics; Longevity; Transcriptome.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. - PubMed
-
- Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14(4):501–513. - PubMed
-
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–198. - PubMed
-
- de Magalhaes JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev. 2018;170:2–9. - PubMed
Publication types
MeSH terms
Grants and funding
- 104978/Z/14/Z/WT_/Wellcome Trust/United Kingdom
- BB/R014949/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- 208375/Z/17/Z/WT_/Wellcome Trust/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- BB/P002579/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
