

AthCNV: A Map of DNA Copy Number Variations in the Arabidopsis Genome
- PMID: 32265262
- PMCID: PMC7268809
- DOI: 10.1105/tpc.19.00640
AthCNV: A Map of DNA Copy Number Variations in the Arabidopsis Genome
Abstract
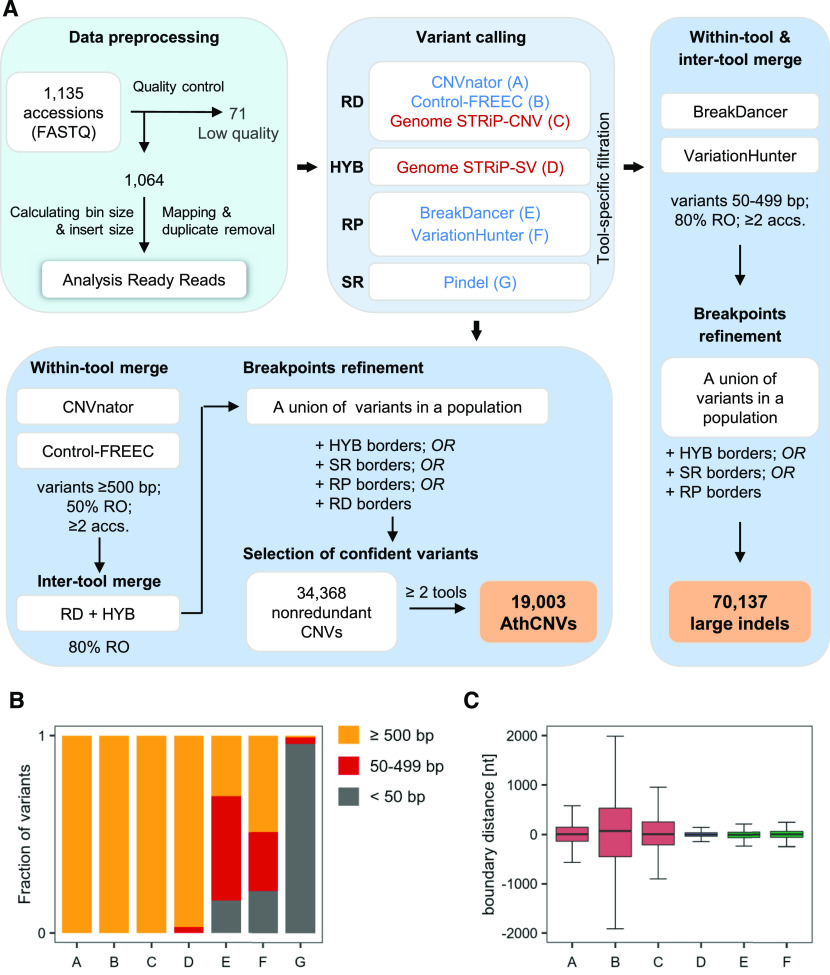
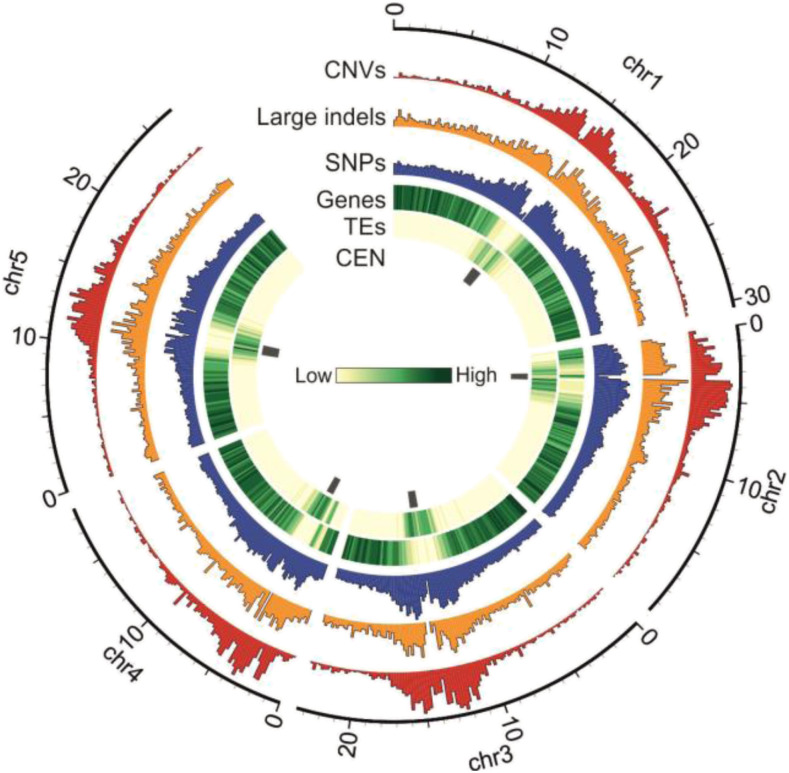
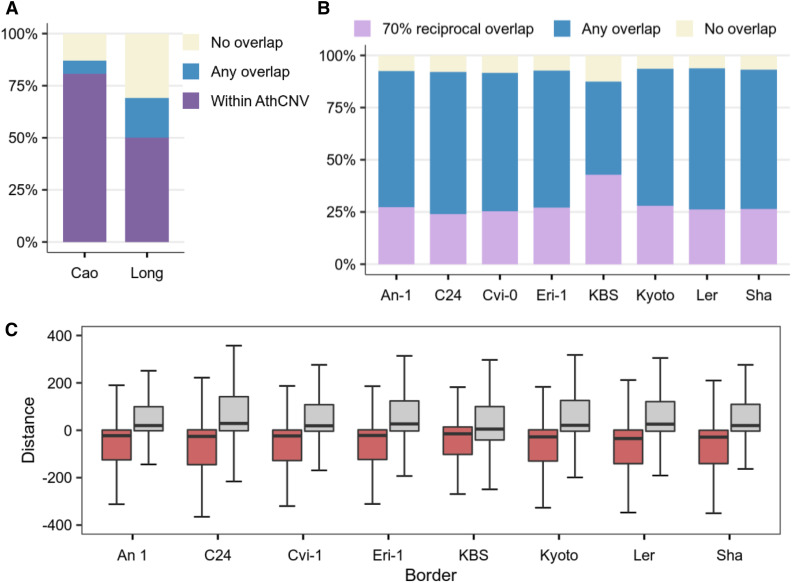
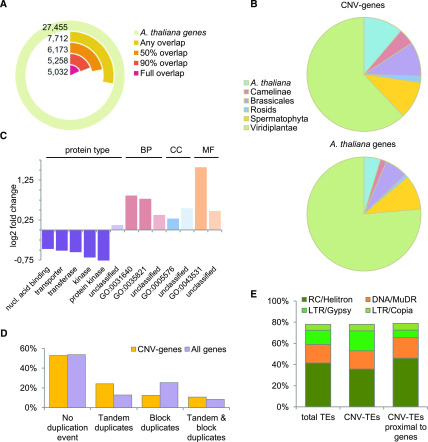
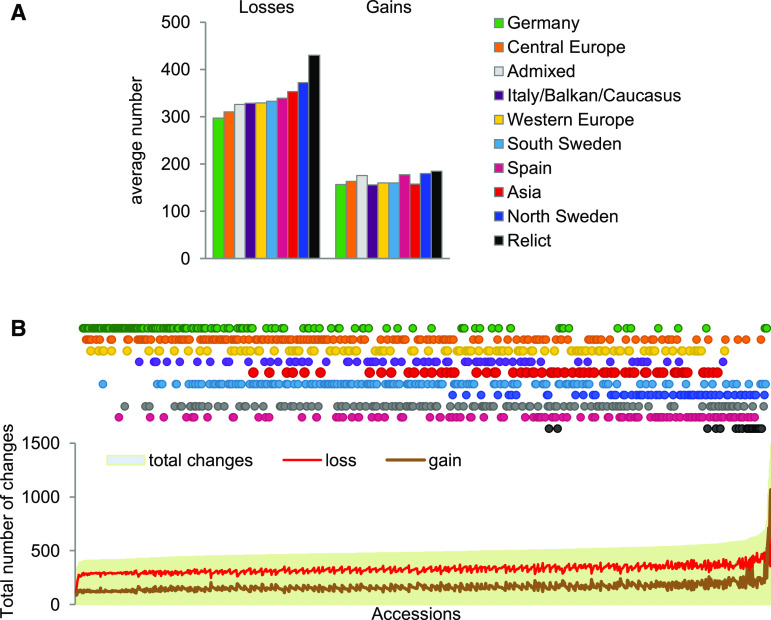
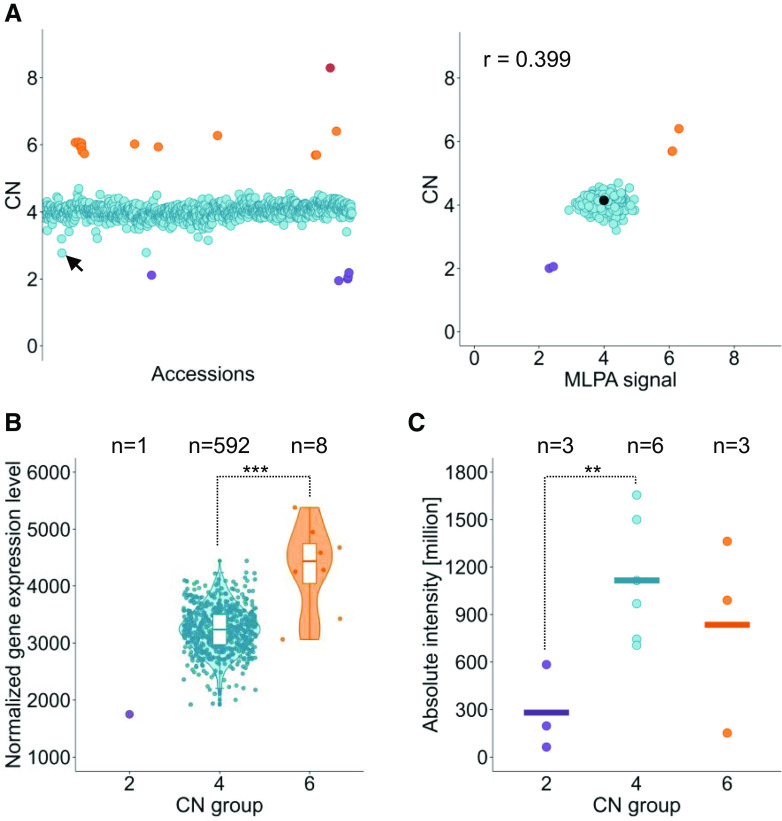
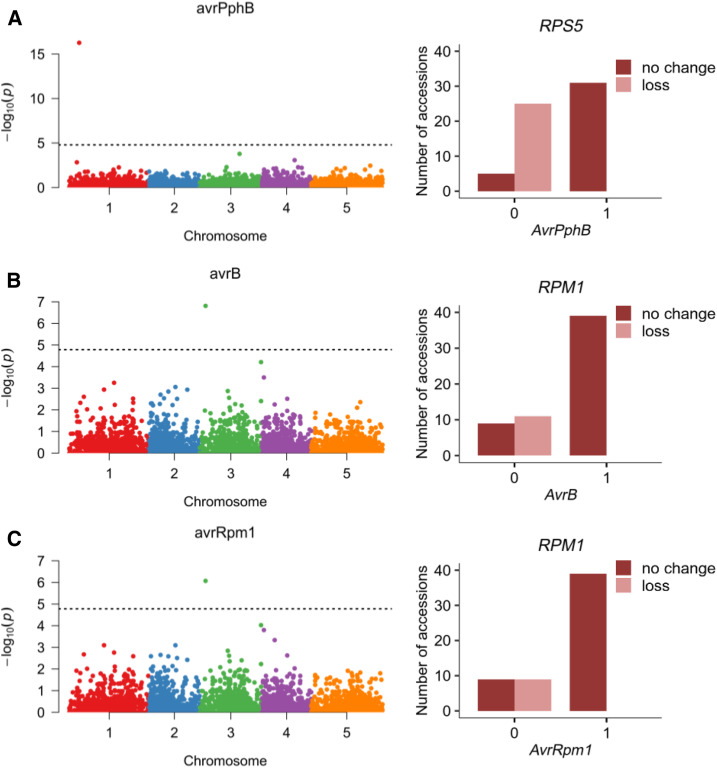
Copy number variations (CNVs) greatly contribute to intraspecies genetic polymorphism and phenotypic diversity. Recent analyses of sequencing data for >1000 Arabidopsis (Arabidopsis thaliana) accessions focused on small variations and did not include CNVs. Here, we performed genome-wide analysis and identified large indels (50 to 499 bp) and CNVs (500 bp and larger) in these accessions. The CNVs fully overlap with 18.3% of protein-coding genes, with enrichment for evolutionarily young genes and genes involved in stress and defense. By combining analysis of both genes and transposable elements (TEs) affected by CNVs, we revealed that the variation statuses of genes and TEs are tightly linked and jointly contribute to the unequal distribution of these elements in the genome. We also determined the gene copy numbers in a set of 1060 accessions and experimentally validated the accuracy of our predictions by multiplex ligation-dependent probe amplification assays. We then successfully used the CNVs as markers to analyze population structure and migration patterns. Finally, we examined the impact of gene dosage variation triggered by a CNV spanning the SEC10 gene on SEC10 expression at both the transcript and protein levels. The catalog of CNVs, CNV-overlapping genes, and their genotypes in a top model dicot will stimulate the exploration of the genetic basis of phenotypic variation.
© 2020 American Society of Plant Biologists. All rights reserved.
Figures
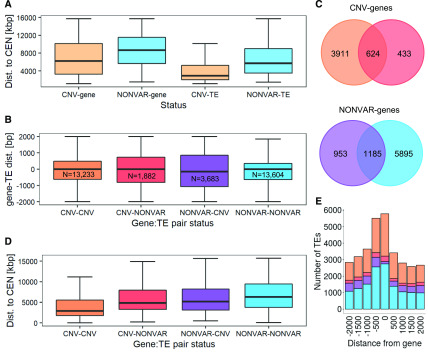
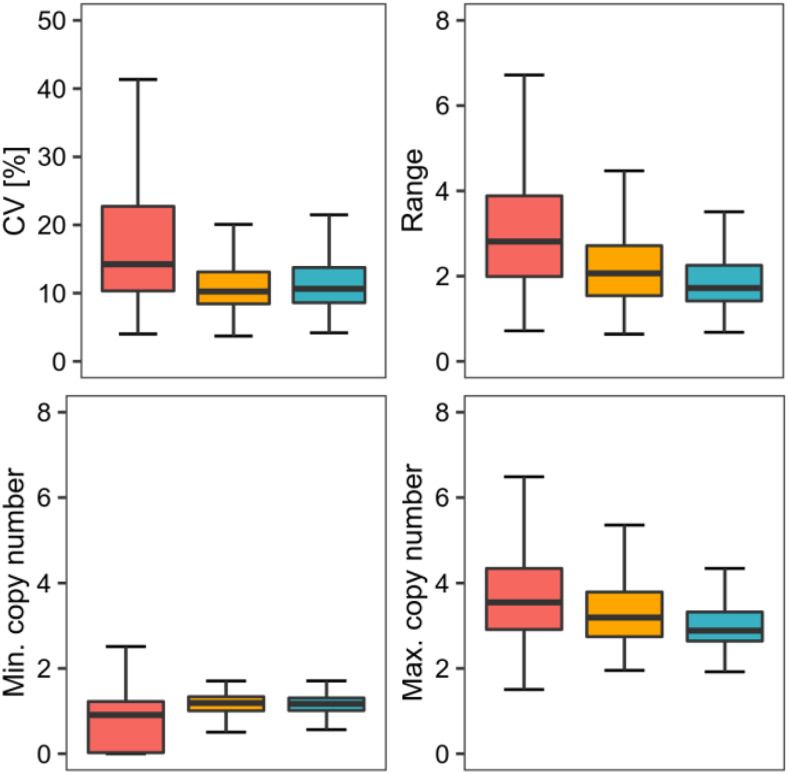
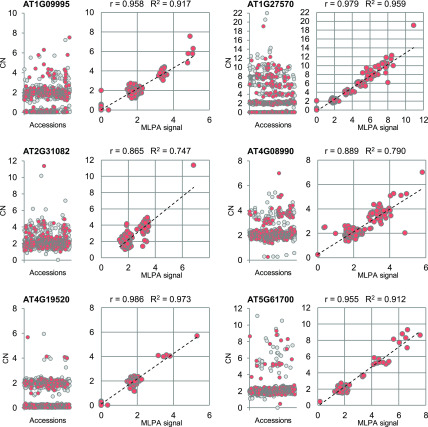
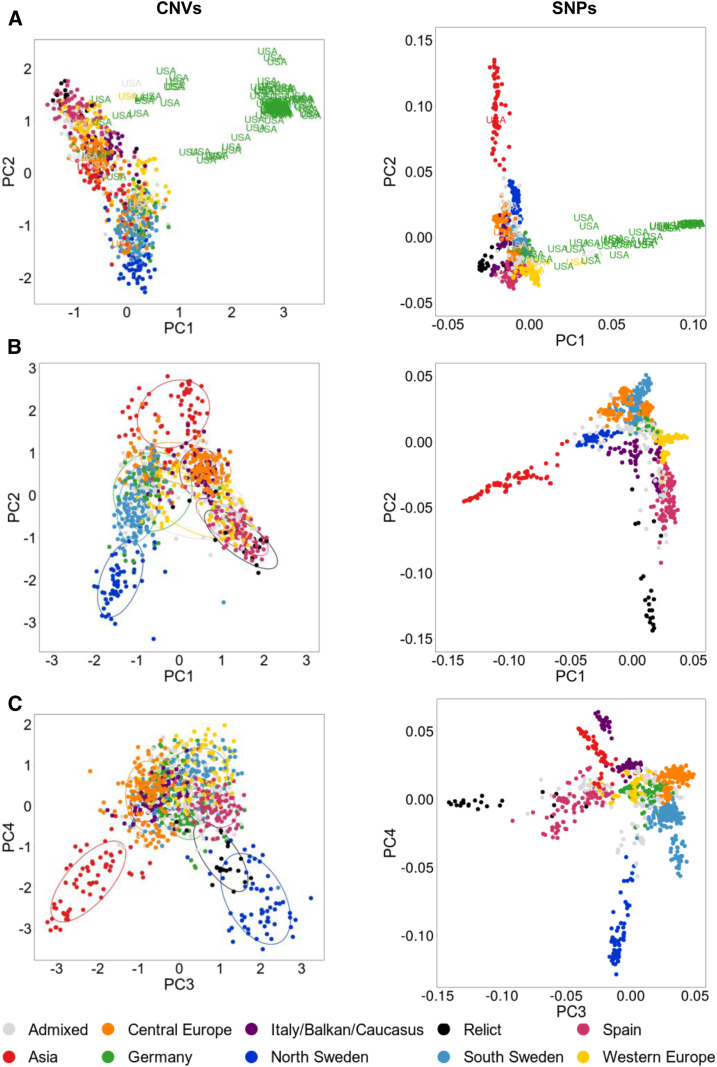

Comment in
-
On the Importance of Variation: A High-Resolution Map of Copy Number Variants in Arabidopsis.Plant Cell. 2020 Jun;32(6):1771-1772. doi: 10.1105/tpc.20.00257. Epub 2020 Apr 7. Plant Cell. 2020. PMID: 32265264 Free PMC article. No abstract available.
References
-
- Alonso-Blanco C., Koornneef M.(2000). Naturally occurring variation in Arabidopsis: An underexploited resource for plant genetics. Trends Plant Sci. 5: 22–29. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
