

A Virus Hosted in Malaria-Infected Blood Protects against T Cell-Mediated Inflammatory Diseases by Impairing DC Function in a Type I IFN-Dependent Manner
- PMID: 32265335
- PMCID: PMC7157782
- DOI: 10.1128/mBio.03394-19
A Virus Hosted in Malaria-Infected Blood Protects against T Cell-Mediated Inflammatory Diseases by Impairing DC Function in a Type I IFN-Dependent Manner
Abstract
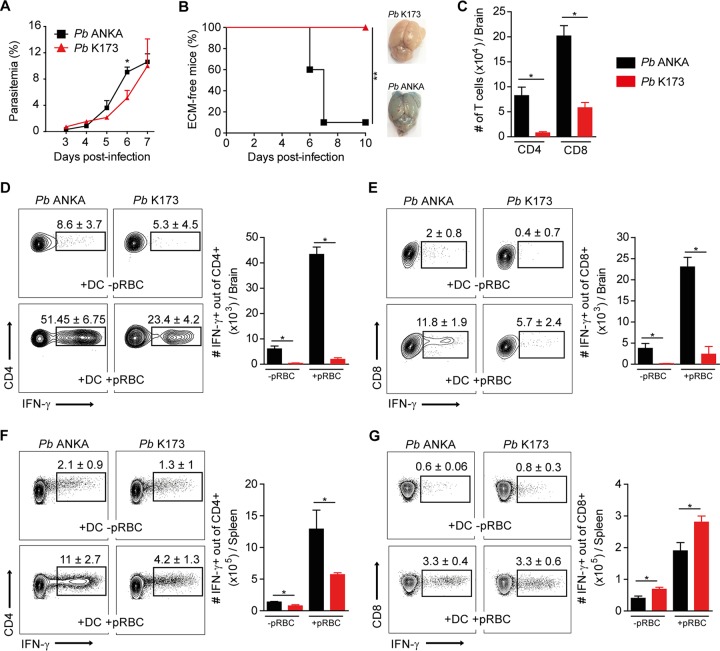
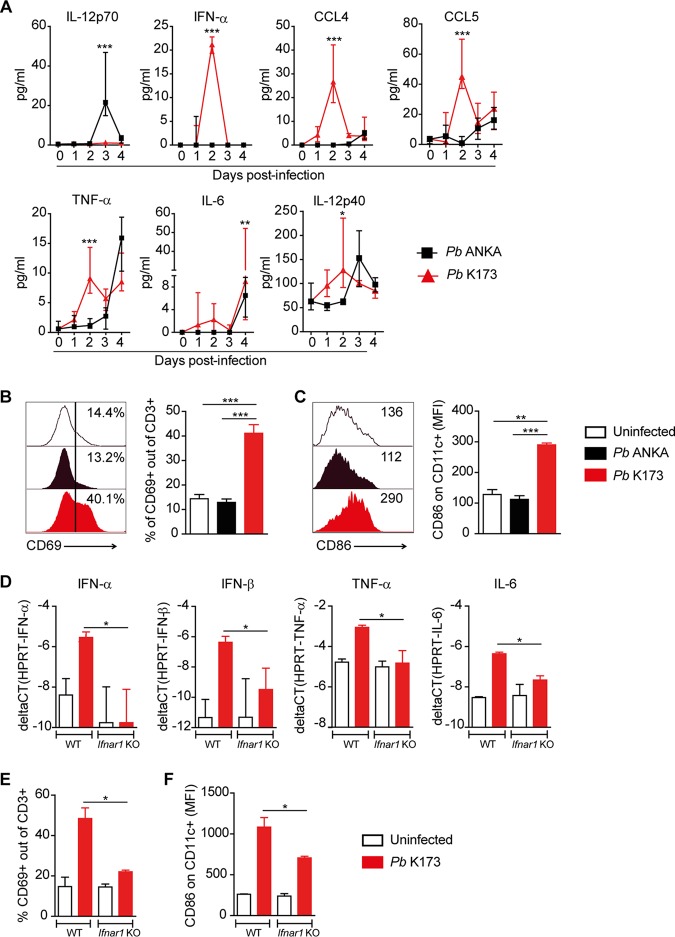
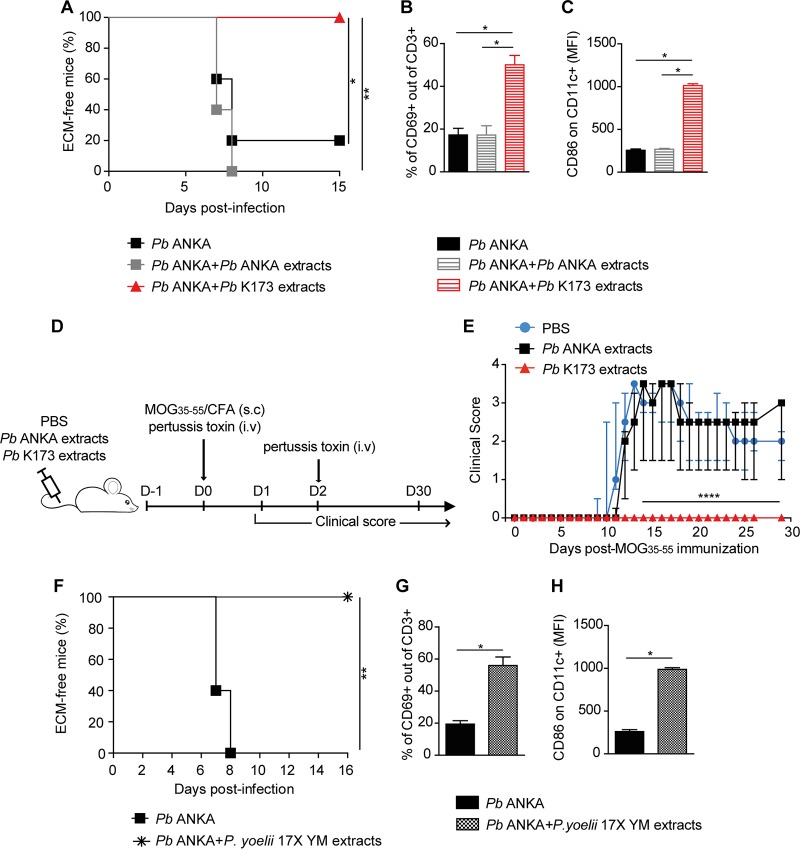
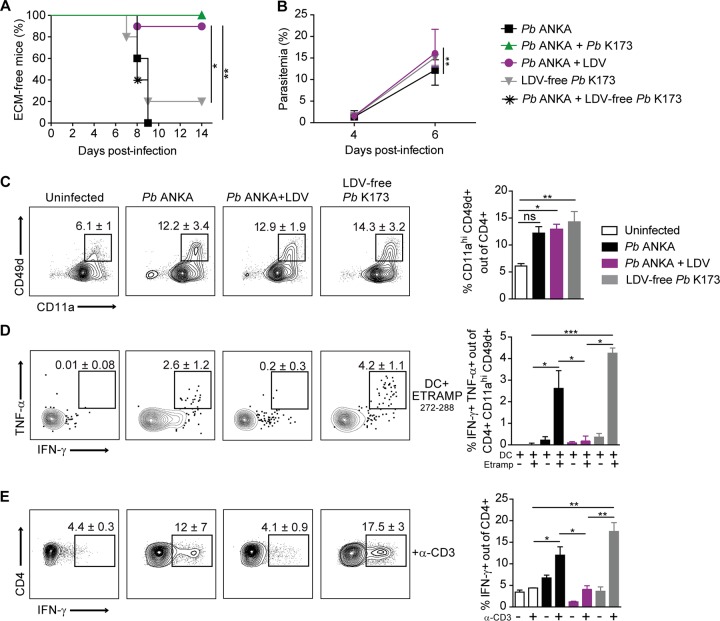
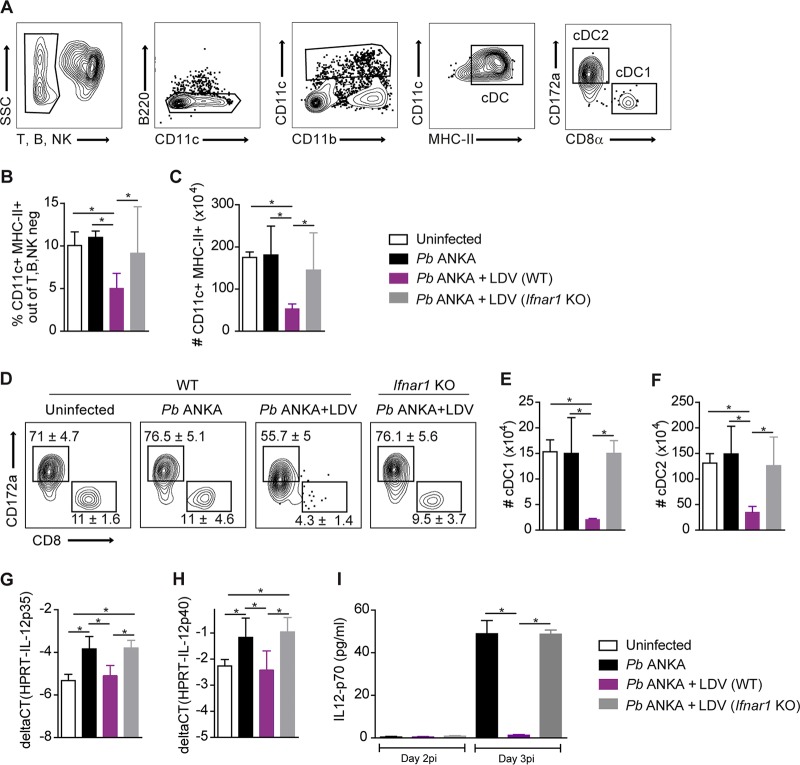
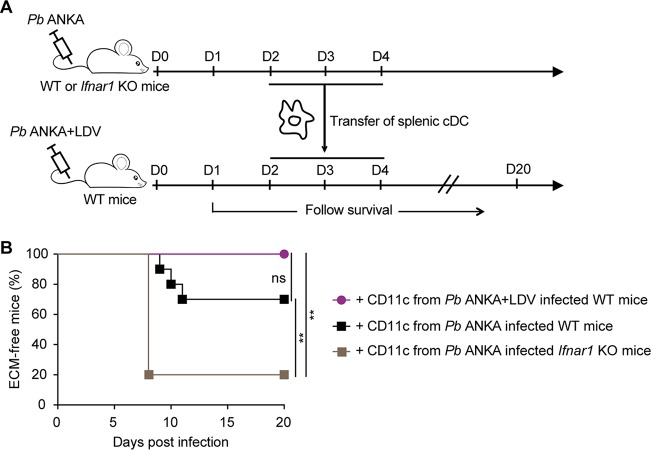
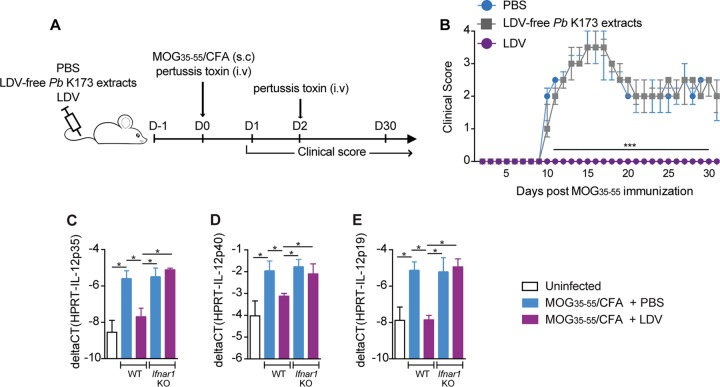
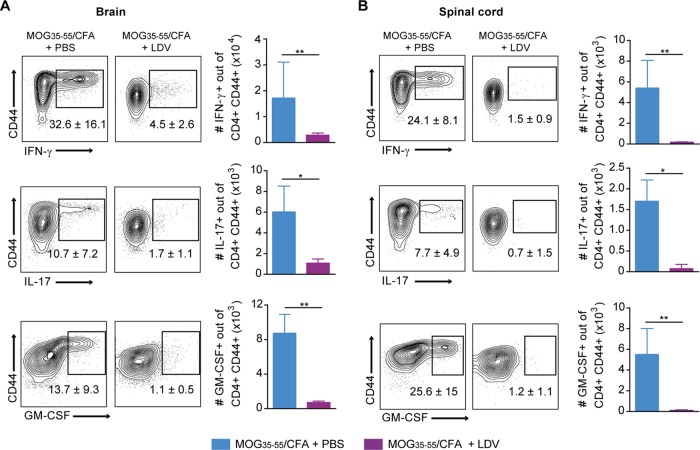
Coinfections shape immunity and influence the development of inflammatory diseases, resulting in detrimental or beneficial outcome. Coinfections with concurrent Plasmodium species can alter malaria clinical evolution, and malaria infection itself can modulate autoimmune reactions. Yet, the underlying mechanisms remain ill defined. Here, we demonstrate that the protective effects of some rodent malaria strains on T cell-mediated inflammatory pathologies are due to an RNA virus cohosted in malaria-parasitized blood. We show that live and extracts of blood parasitized by Plasmodium berghei K173 or Plasmodium yoelii 17X YM, protect against P. berghei ANKA-induced experimental cerebral malaria (ECM) and myelin oligodendrocyte glycoprotein (MOG)/complete Freund's adjuvant (CFA)-induced experimental autoimmune encephalomyelitis (EAE), and that protection is associated with a strong type I interferon (IFN-I) signature. We detected the presence of the RNA virus lactate dehydrogenase-elevating virus (LDV) in the protective Plasmodium stabilates and we established that LDV infection alone was necessary and sufficient to recapitulate the protective effects on ECM and EAE. In ECM, protection resulted from an IFN-I-mediated reduction in the abundance of splenic conventional dendritic cell and impairment of their ability to produce interleukin (IL)-12p70, leading to a decrease in pathogenic CD4+ Th1 responses. In EAE, LDV infection induced IFN-I-mediated abrogation of IL-23, thereby preventing the differentiation of granulocyte-macrophage colony-stimulating factor (GM-CSF)-producing encephalitogenic CD4+ T cells. Our work identifies a virus cohosted in several Plasmodium stabilates across the community and deciphers its major consequences on the host immune system. More generally, our data emphasize the importance of considering contemporaneous infections for the understanding of malaria-associated and autoimmune diseases.IMPORTANCE Any infection modifies the host immune status, potentially ameliorating or aggravating the pathophysiology of a simultaneous inflammatory condition. In the course of investigating how malaria infection modulates the severity of contemporaneous inflammatory diseases, we identified a nonpathogenic mouse virus in stabilates of two widely used rodent parasite lines: Plasmodium berghei K173 and Plasmodium yoelii 17X YM. We established that the protective effects of these Plasmodium lines on cerebral malaria and multiple sclerosis are exclusively due to this virus. The virus induces a massive type I interferon (IFN-I) response and causes quantitative and qualitative defects in the ability of dendritic cells to promote pathogenic T cell responses. Beyond revealing a possible confounding factor in rodent malaria models, our work uncovers some bases by which a seemingly innocuous viral (co)infection profoundly changes the immunopathophysiology of inflammatory diseases.
Keywords: CD4 T cell; RNA virus; autoimmunity; coinfection; dendritic cells; inflammation; malaria.
Copyright © 2020 Hassan et al.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
