

Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2'-O-ribose methyltransferase
- PMID: 32266873
- PMCID: PMC7189412
- DOI: 10.1080/07391102.2020.1753577
Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2'-O-ribose methyltransferase
Abstract
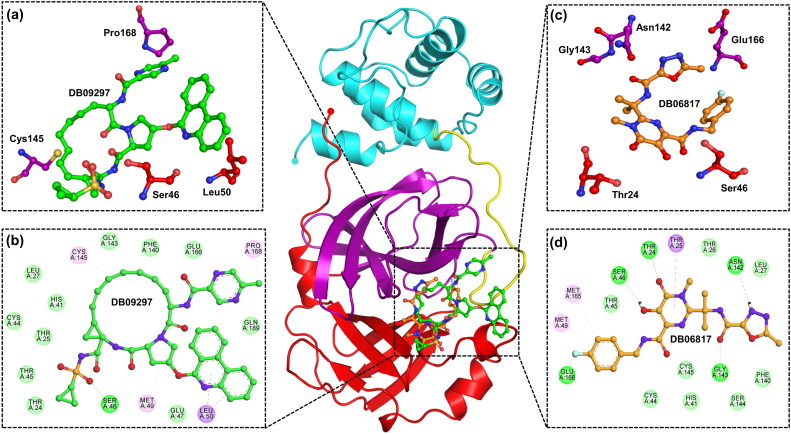
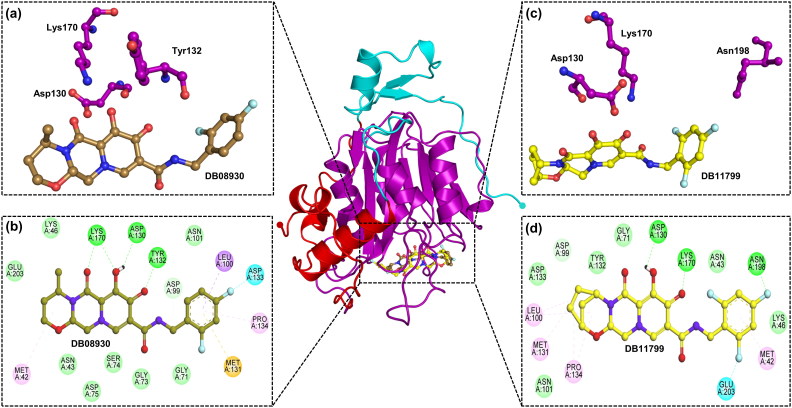
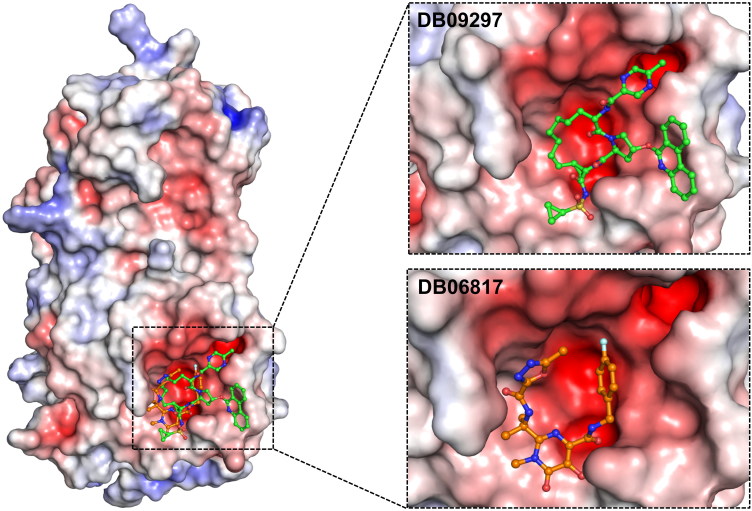
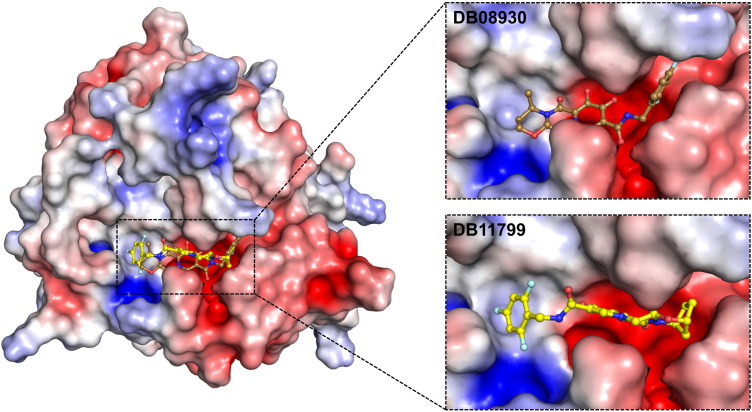
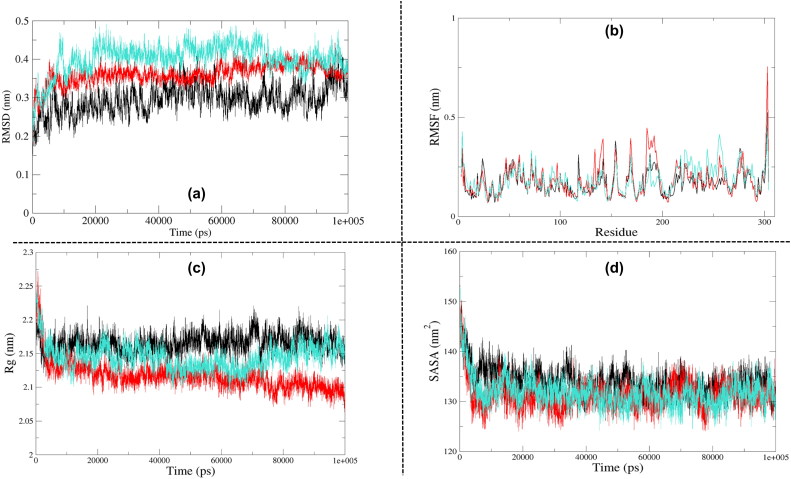
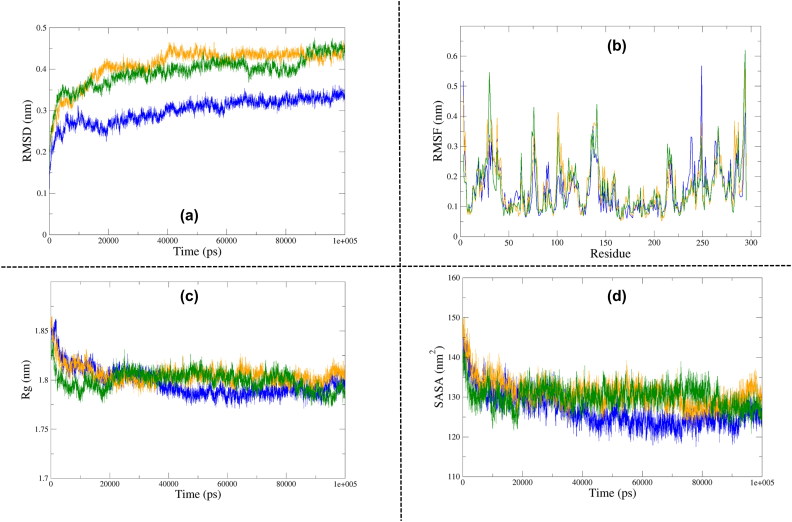
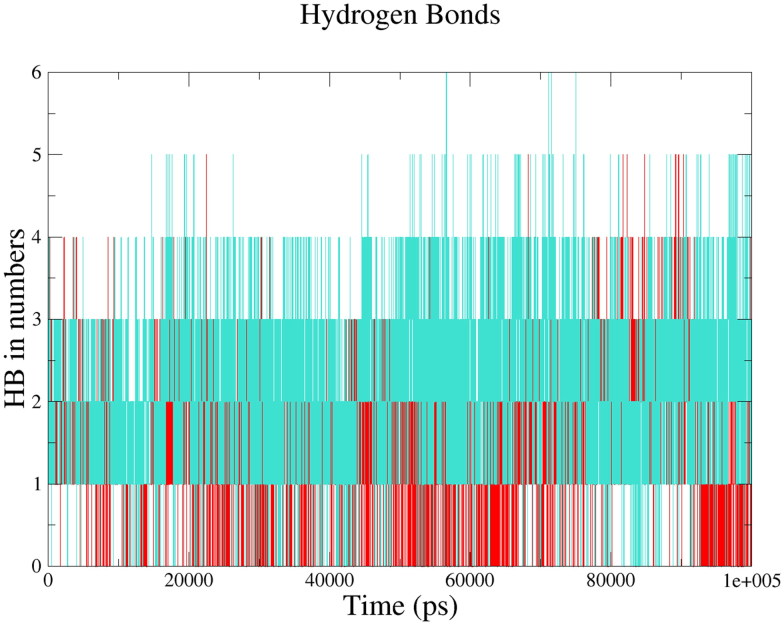
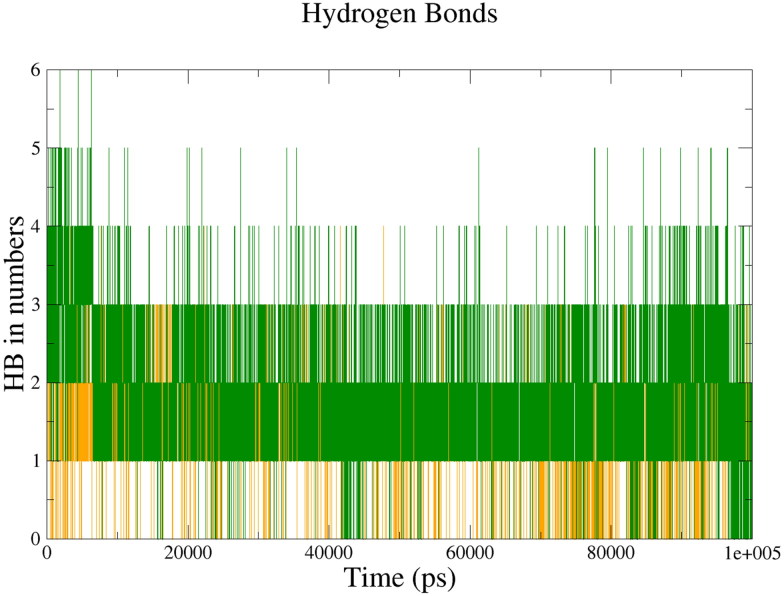
The recent pandemic associated with SARS-CoV-2, a virus of the Coronaviridae family, has resulted in an unprecedented number of infected people. The highly contagious nature of this virus makes it imperative for us to identify promising inhibitors from pre-existing antiviral drugs. Two druggable targets, namely 3C-like proteinase (3CLpro) and 2'-O-ribose methyltransferase (2'-O-MTase) were selected in this study due to their indispensable nature in the viral life cycle. 3CLpro is a cysteine protease responsible for the proteolysis of replicase polyproteins resulting in the formation of various functional proteins, whereas 2'-O-MTase methylates the ribose 2'-O position of the first and second nucleotide of viral mRNA, which sequesters it from the host immune system. The selected drug target proteins were screened against an in-house library of 123 antiviral drugs. Two promising drug molecules were identified for each protein based on their estimated free energy of binding (ΔG), the orientation of drug molecules in the active site and the interacting residues. The selected protein-drug complexes were then subjected to MD simulation, which consists of various structural parameters to equivalently reflect their physiological state. From the virtual screening results, two drug molecules were selected for each drug target protein [Paritaprevir (ΔG = -9.8 kcal/mol) & Raltegravir (ΔG = -7.8 kcal/mol) for 3CLpro and Dolutegravir (ΔG = -9.4 kcal/mol) and Bictegravir (ΔG = -8.4 kcal/mol) for 2'-OMTase]. After the extensive computational analysis, we proposed that Raltegravir, Paritaprevir, Bictegravir and Dolutegravir are excellent lead candidates for these crucial proteins and they could become potential therapeutic drugs against SARS-CoV-2. Communicated by Ramaswamy H. Sarma.
Keywords: 2′-O-ribose methyltransferase; 3C-like proteinase; Drug repurposing; MD simulation; SARS-CoV-2; docking.
Figures
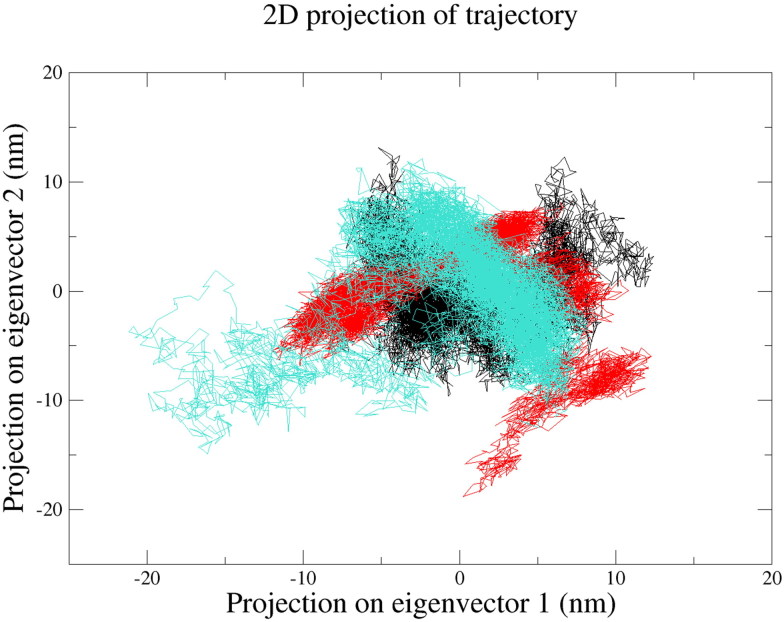
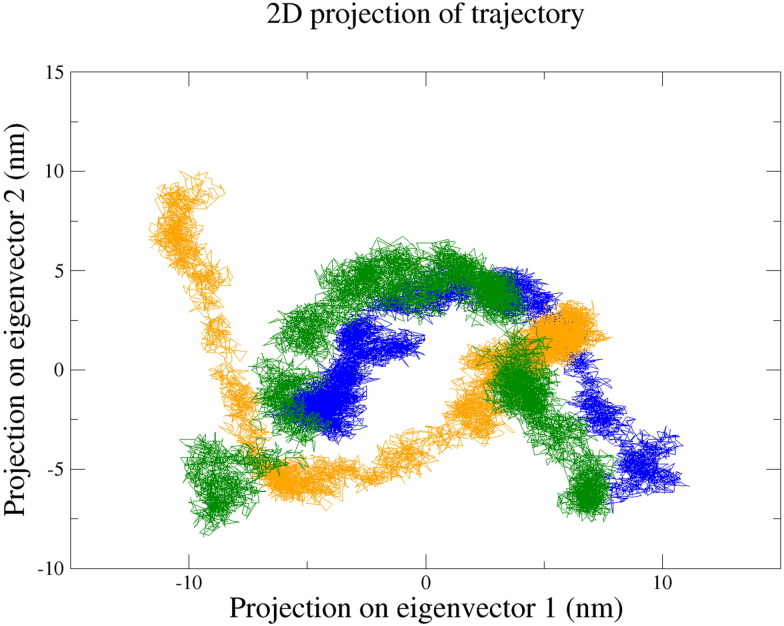
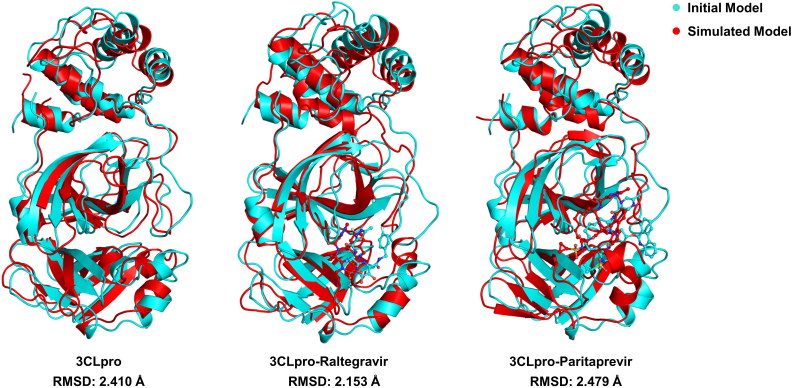
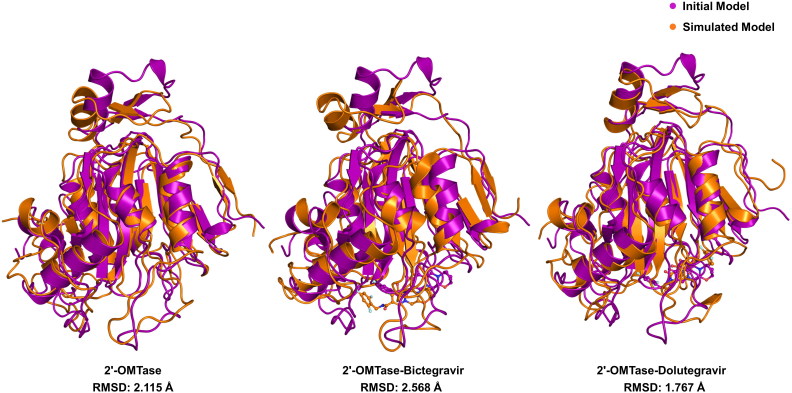
References
-
- Abraham M. J., Lindahl D., van der Spoel E., Hess B., & the GROMACS development team (2014). GROMACS User Manual version, 5(2), 1–298.
-
- Amera G. M., Khan R. J., Pathak A., Jha R. K., Muthukumaran J., & Singh A. K. (2019). Screening of promising molecules against MurG as drug target in multi-drug-resistant-Acinetobacter baumannii – Insights from comparative protein modeling, molecular docking and molecular dynamics simulation. Journal of Biomolecular Structure and Dynamics, 1–23. doi:10.1080/07391102.2019.1700167 - DOI - PubMed
-
- Amera G. M., Khan R. J., Pathak A., Kumar A., & Singh A. K. (2019). Structure based in-silico study on UDP-N-acetylmuramoyl-L-alanyl-D-glutamate-2,6-diaminopimelate ligase (MurE) from Acinetobacter baumannii as a drug target against nosocomial infections. Informatics in Medicine Unlocked, 16, 100216. doi:10.1016/j.imu.2019.100216 - DOI
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous