

Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex
- PMID: 32268104
- PMCID: PMC7295160
- DOI: 10.1016/j.celrep.2020.03.053
Whole-Genome and RNA Sequencing Reveal Variation and Transcriptomic Coordination in the Developing Human Prefrontal Cortex
Abstract
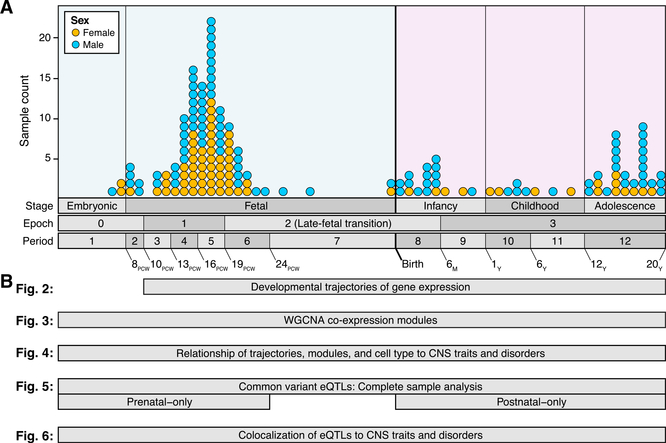
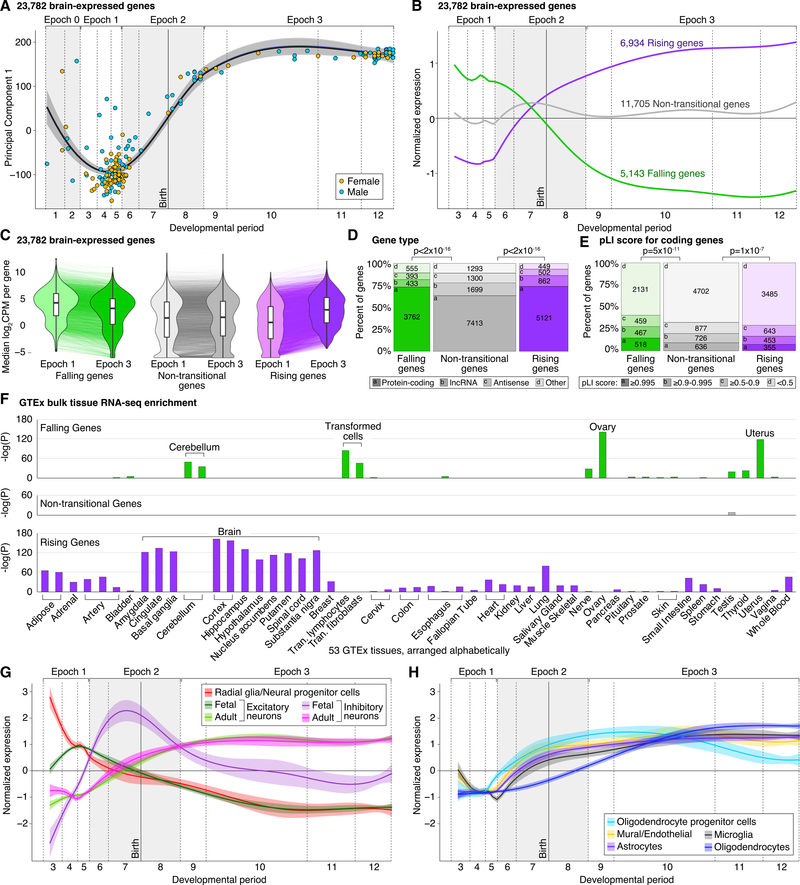
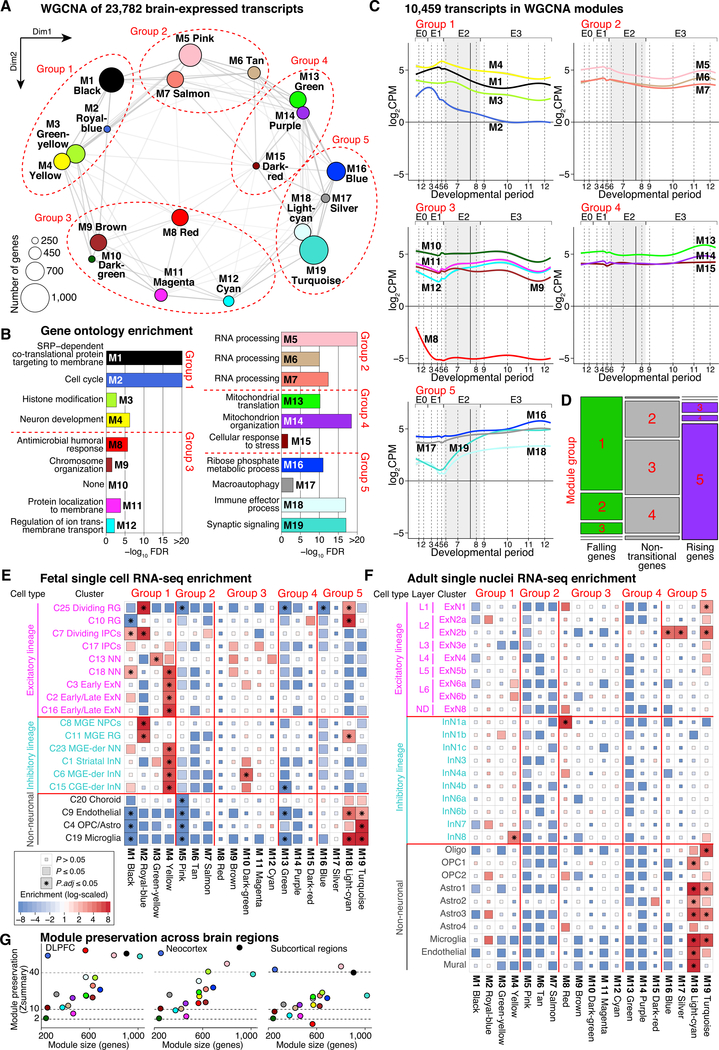
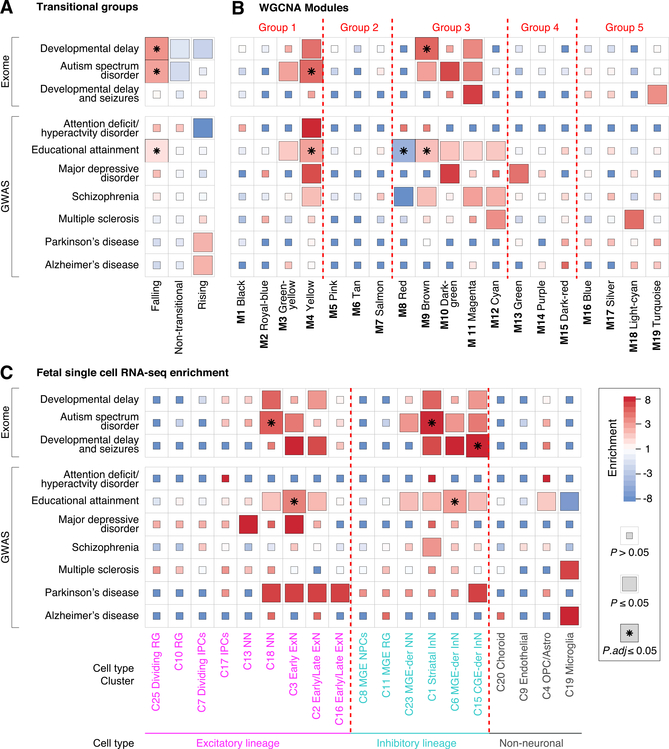
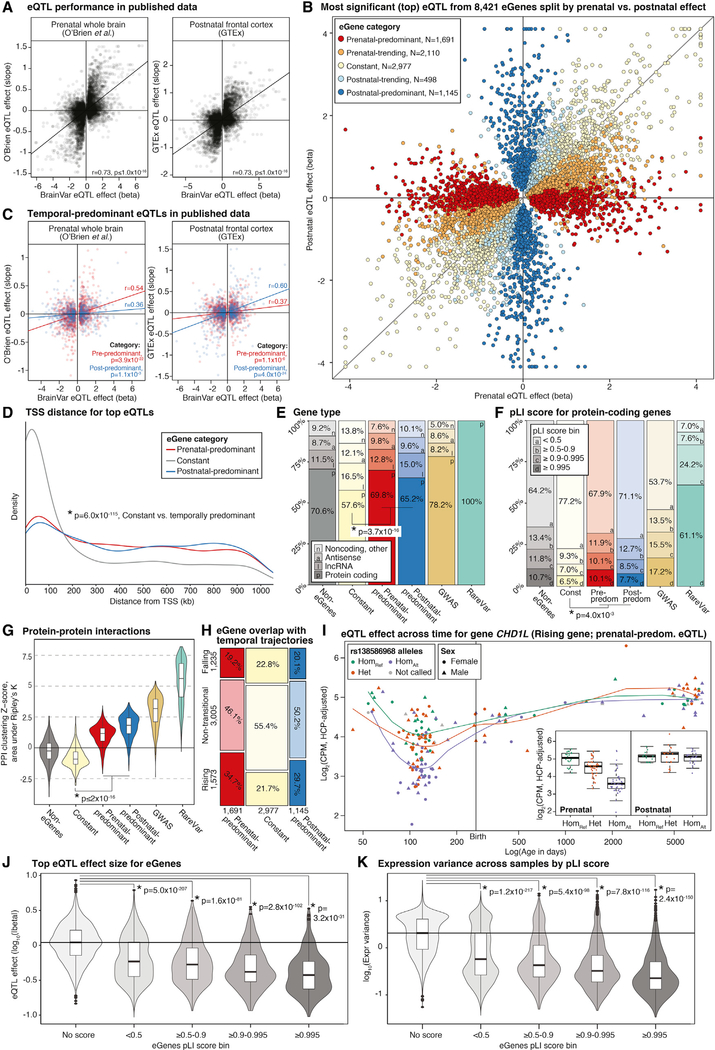
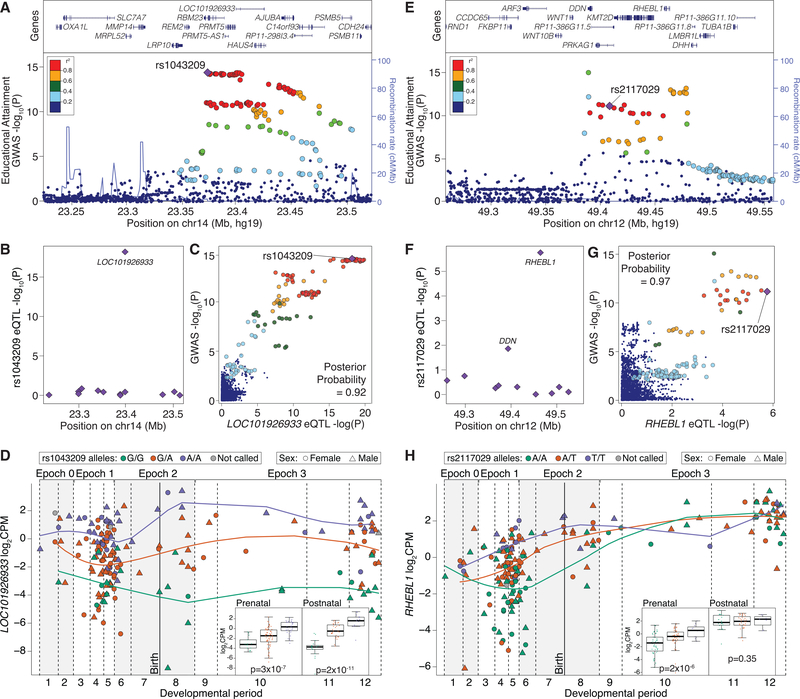
Gene expression levels vary across developmental stage, cell type, and region in the brain. Genomic variants also contribute to the variation in expression, and some neuropsychiatric disorder loci may exert their effects through this mechanism. To investigate these relationships, we present BrainVar, a unique resource of paired whole-genome and bulk tissue RNA sequencing from the dorsolateral prefrontal cortex of 176 individuals across prenatal and postnatal development. Here we identify common variants that alter gene expression (expression quantitative trait loci [eQTLs]) constantly across development or predominantly during prenatal or postnatal stages. Both "constant" and "temporal-predominant" eQTLs are enriched for loci associated with neuropsychiatric traits and disorders and colocalize with specific variants. Expression levels of more than 12,000 genes rise or fall in a concerted late-fetal transition, with the transitional genes enriched for cell-type-specific genes and neuropsychiatric risk loci, underscoring the importance of cataloging developmental trajectories in understanding cortical physiology and pathology.
Keywords: BrainVar; DLPFC; LOC101926933 RP11-298I3.1 AL132780.1 ENSG00000257285; PsychENCODE; RHEBL1; dorsolateral prefrontal cortex; fetal transition; mTOR; prenatal eQTL.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Bae BI, and Walsh CA (2013). Neuroscience. What are mini-brains? Science 342, 200–201.
Publication types
MeSH terms
Grants and funding
- R01 MH110928/MH/NIMH NIH HHS/United States
- R01 MH094714/MH/NIMH NIH HHS/United States
- R21 MH103877/MH/NIMH NIH HHS/United States
- U01 MH103365/MH/NIMH NIH HHS/United States
- R03 DE025665/DE/NIDCR NIH HHS/United States
- R01 HG006399/HG/NHGRI NIH HHS/United States
- U01 MH103392/MH/NIMH NIH HHS/United States
- R01 MH057881/MH/NIMH NIH HHS/United States
- U01 MH103346/MH/NIMH NIH HHS/United States
- R01 MH105472/MH/NIMH NIH HHS/United States
- U01 MH103340/MH/NIMH NIH HHS/United States
- U01 MH103339/MH/NIMH NIH HHS/United States
- R01 MH109904/MH/NIMH NIH HHS/United States
- R21 MH105881/MH/NIMH NIH HHS/United States
- U01 MH106874/MH/NIMH NIH HHS/United States
- R01 MH110926/MH/NIMH NIH HHS/United States
- U01 MH116488/MH/NIMH NIH HHS/United States
- P50 MH106934/MH/NIMH NIH HHS/United States
- R01 MH115957/MH/NIMH NIH HHS/United States
- R37 MH057881/MH/NIMH NIH HHS/United States
- U01 MH105575/MH/NIMH NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- R01 CA227237/CA/NCI NIH HHS/United States
- R01 MH109901/MH/NIMH NIH HHS/United States
- R01 HD081256/HD/NICHD NIH HHS/United States
- R24 HD000836/HD/NICHD NIH HHS/United States
- U01 HG009080/HG/NHGRI NIH HHS/United States
- U01 MH111662/MH/NIMH NIH HHS/United States
- R21 MH102791/MH/NIMH NIH HHS/United States
- R01 MH105898/MH/NIMH NIH HHS/United States
- R01 HD096326/HD/NICHD NIH HHS/United States
- K25 HL121295/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
