

Consensus guidelines for management of hyperammonaemia in paediatric patients receiving continuous kidney replacement therapy
- PMID: 32269302
- PMCID: PMC7366888
- DOI: 10.1038/s41581-020-0267-8
Consensus guidelines for management of hyperammonaemia in paediatric patients receiving continuous kidney replacement therapy
Abstract
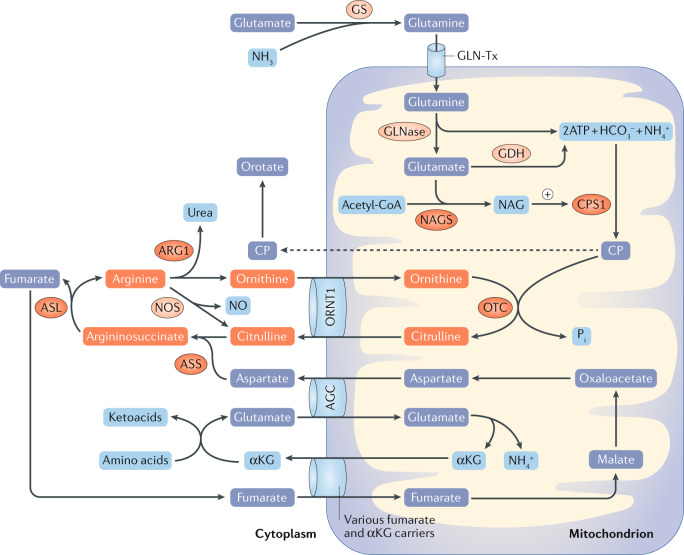
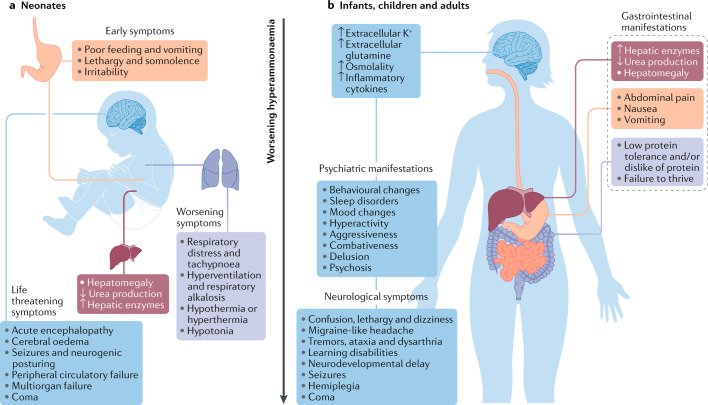
Hyperammonaemia in children can lead to grave consequences in the form of cerebral oedema, severe neurological impairment and even death. In infants and children, common causes of hyperammonaemia include urea cycle disorders or organic acidaemias. Few studies have assessed the role of extracorporeal therapies in the management of hyperammonaemia in neonates and children. Moreover, consensus guidelines are lacking for the use of non-kidney replacement therapy (NKRT) and kidney replacement therapies (KRTs, including peritoneal dialysis, continuous KRT, haemodialysis and hybrid therapy) to manage hyperammonaemia in neonates and children. Prompt treatment with KRT and/or NKRT, the choice of which depends on the ammonia concentrations and presenting symptoms of the patient, is crucial. This expert Consensus Statement presents recommendations for the management of hyperammonaemia requiring KRT in paediatric populations. Additional studies are required to strengthen these recommendations.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Auron A, Brophy PD. Hyperammonemia in review: pathophysiology, diagnosis, and treatment. Pediatr. Nephrol. 2012;27:207–222. - PubMed
-
- Mew, N. A., Pappa, M. B., Gropman, A. L. in Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease (eds Rosenberg, R. N. & Pascual, J. M.) 633–647 (Elsevier, 2014).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
