

Integrating genomic features for non-invasive early lung cancer detection
- PMID: 32269342
- PMCID: PMC8230734
- DOI: 10.1038/s41586-020-2140-0
Integrating genomic features for non-invasive early lung cancer detection
Abstract
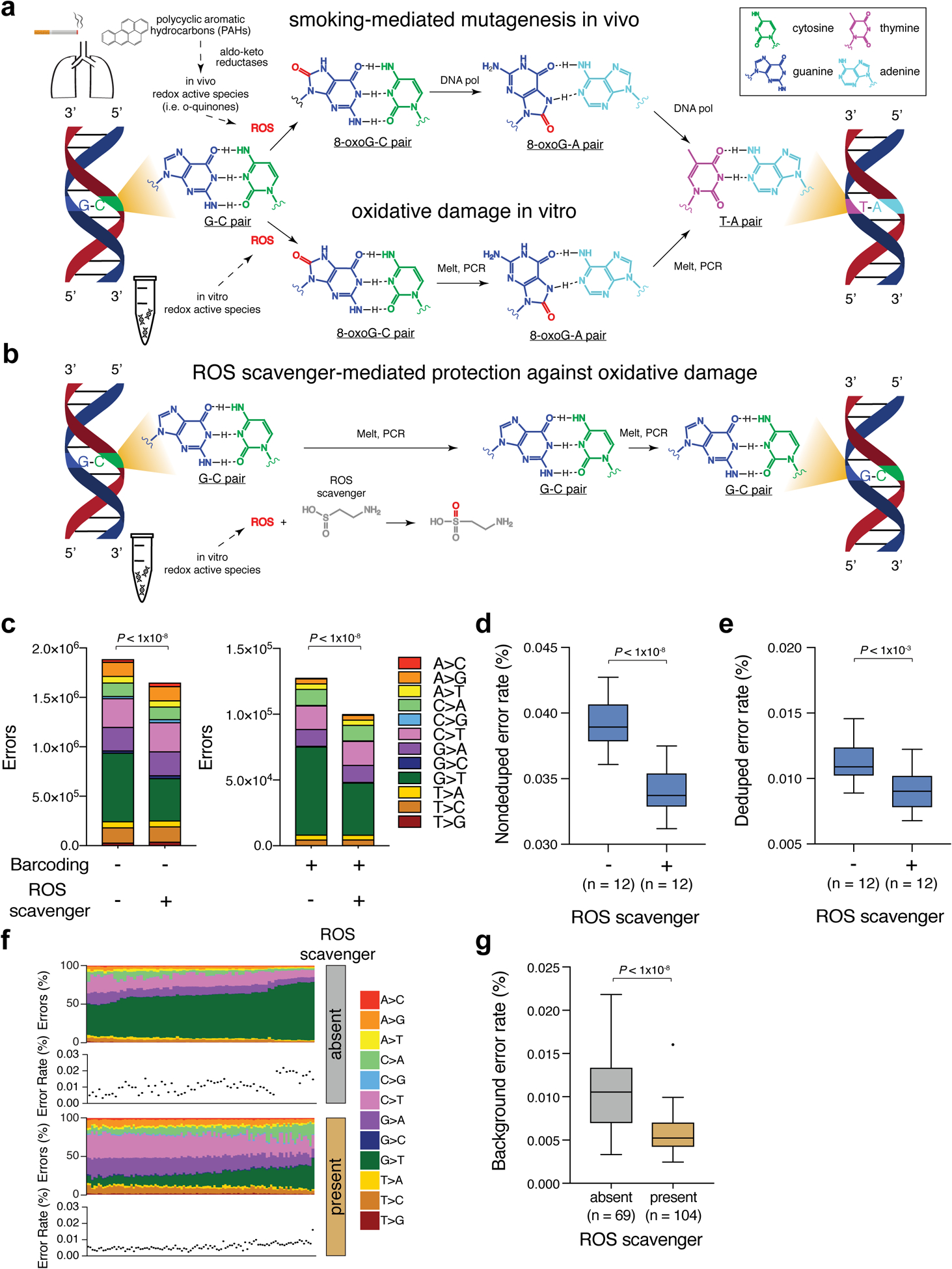
Radiologic screening of high-risk adults reduces lung-cancer-related mortality1,2; however, a small minority of eligible individuals undergo such screening in the United States3,4. The availability of blood-based tests could increase screening uptake. Here we introduce improvements to cancer personalized profiling by deep sequencing (CAPP-Seq)5, a method for the analysis of circulating tumour DNA (ctDNA), to better facilitate screening applications. We show that, although levels are very low in early-stage lung cancers, ctDNA is present prior to treatment in most patients and its presence is strongly prognostic. We also find that the majority of somatic mutations in the cell-free DNA (cfDNA) of patients with lung cancer and of risk-matched controls reflect clonal haematopoiesis and are non-recurrent. Compared with tumour-derived mutations, clonal haematopoiesis mutations occur on longer cfDNA fragments and lack mutational signatures that are associated with tobacco smoking. Integrating these findings with other molecular features, we develop and prospectively validate a machine-learning method termed 'lung cancer likelihood in plasma' (Lung-CLiP), which can robustly discriminate early-stage lung cancer patients from risk-matched controls. This approach achieves performance similar to that of tumour-informed ctDNA detection and enables tuning of assay specificity in order to facilitate distinct clinical applications. Our findings establish the potential of cfDNA for lung cancer screening and highlight the importance of risk-matching cases and controls in cfDNA-based screening studies.
Figures













Comment in
-
Machine Learning Yields Lung Cancer Test.Cancer Discov. 2020 Jun;10(6):753-754. doi: 10.1158/2159-8290.CD-NB2020-033. Epub 2020 Apr 27. Cancer Discov. 2020. PMID: 32341019
-
Liquid biopsy for early stage lung cancer moves ever closer.Nat Rev Clin Oncol. 2020 Sep;17(9):523-524. doi: 10.1038/s41571-020-0393-z. Nat Rev Clin Oncol. 2020. PMID: 32457540 No abstract available.
-
Oncology Scan: Radiation Biology and Genomic Predictors of Response.Int J Radiat Oncol Biol Phys. 2020 Jul 1;107(3):393-397. doi: 10.1016/j.ijrobp.2020.04.008. Int J Radiat Oncol Biol Phys. 2020. PMID: 32531379 Free PMC article. No abstract available.
References
-
- Moyer VA Screening for Lung Cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann. Intern. Med 160, 330–338 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- DP2 CA186569/CA/NCI NIH HHS/United States
- T32 CA009302/CA/NCI NIH HHS/United States
- R25 CA180993/CA/NCI NIH HHS/United States
- T32 CA121940/CA/NCI NIH HHS/United States
- K12 CA090628/CA/NCI NIH HHS/United States
- P30 CA015083-44S1/CA/NCI NIH HHS/United States
- R01 CA233975/CA/NCI NIH HHS/United States
- P30 CA015083/CA/NCI NIH HHS/United States
- K08 CA241076/CA/NCI NIH HHS/United States
- 1-K08-CA241076-01/CA/NCI NIH HHS/United States
- U01 CA196405/CA/NCI NIH HHS/United States
- R01 CA188298/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
