

Guidelines on clinical presentation and management of nondystrophic myotonias
- PMID: 32270509
- PMCID: PMC8117169
- DOI: 10.1002/mus.26887
Guidelines on clinical presentation and management of nondystrophic myotonias
Abstract
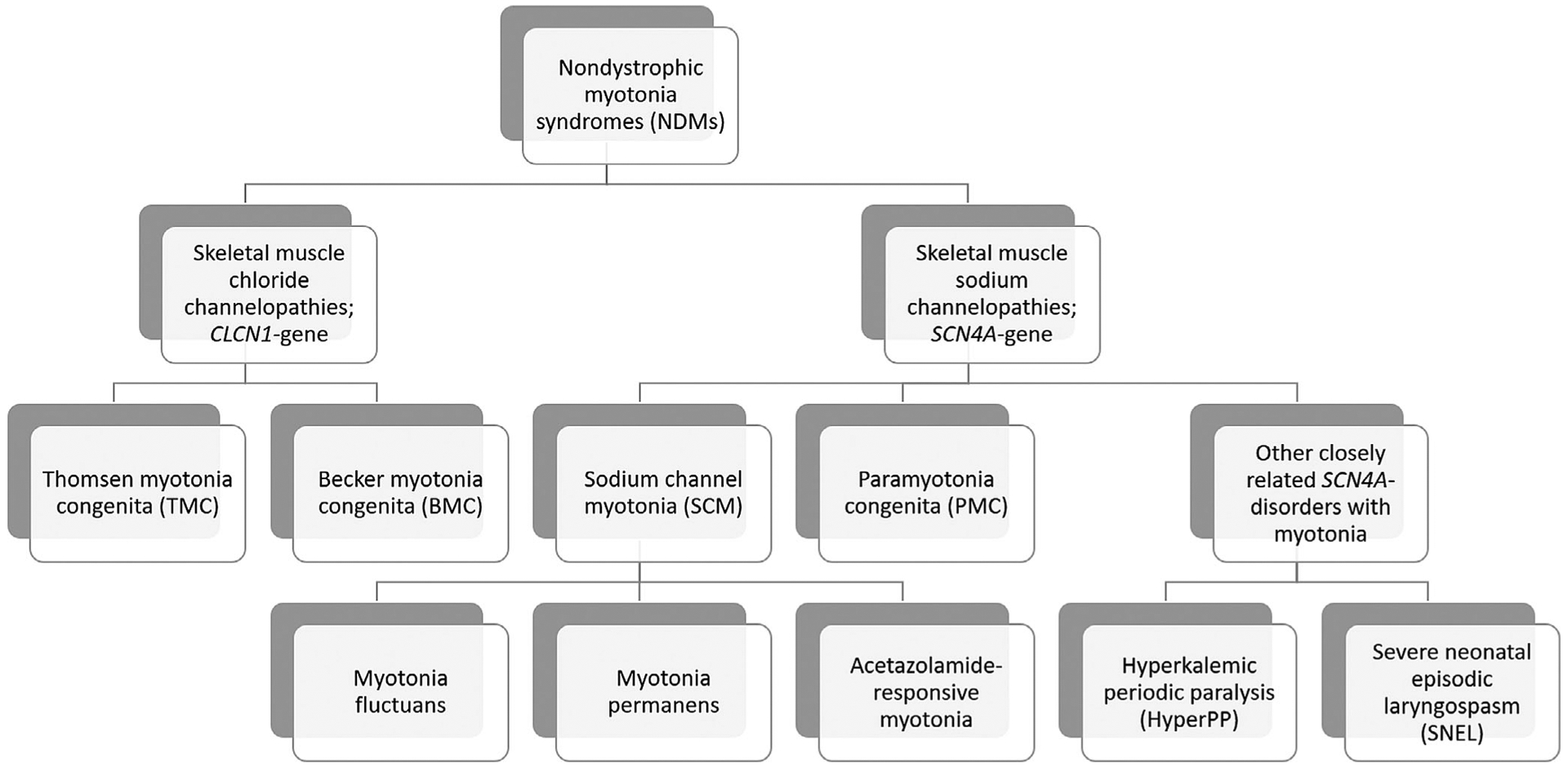
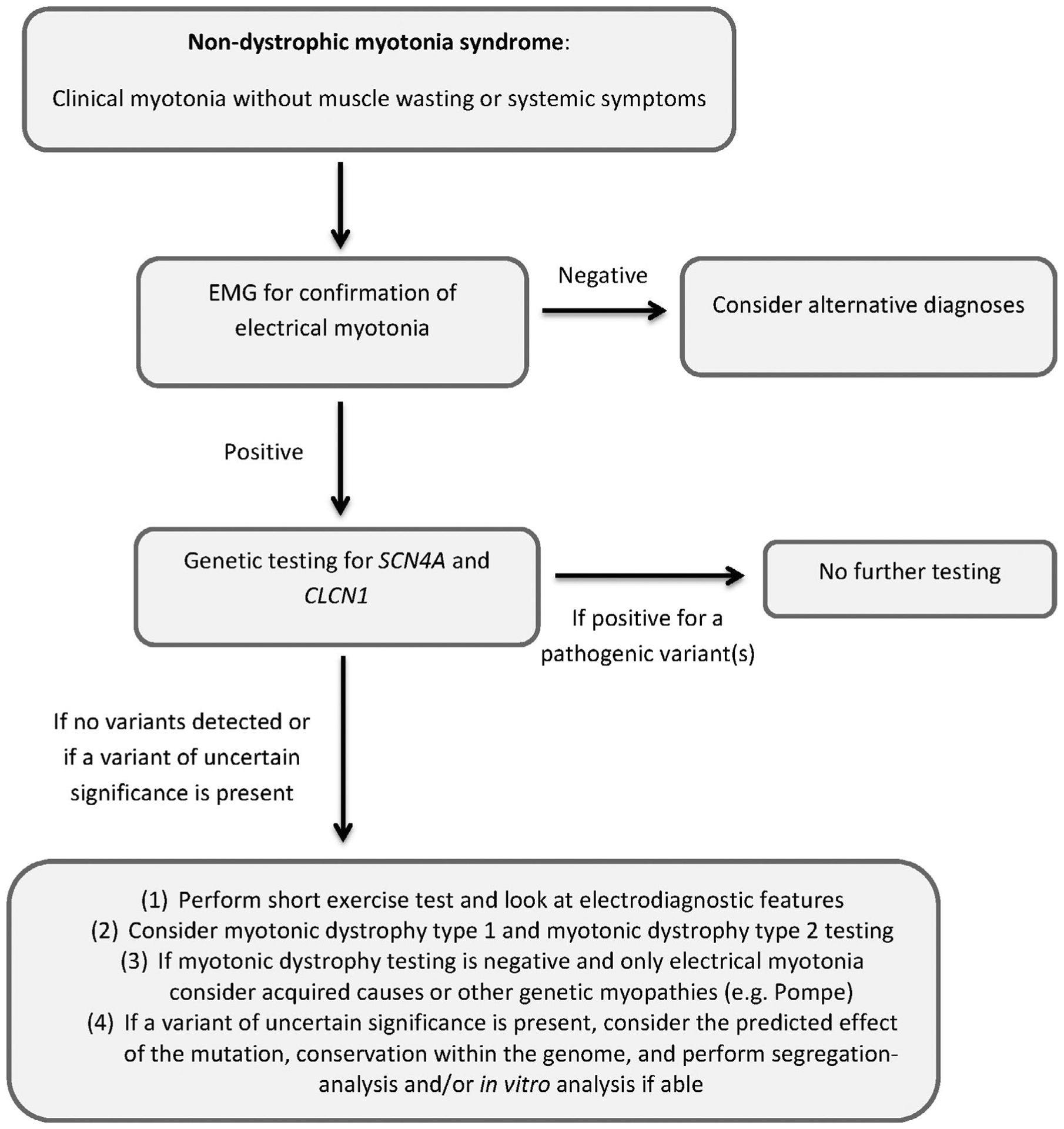
The nondystrophic myotonias are rare muscle hyperexcitability disorders caused by gain-of-function mutations in the SCN4A gene or loss-of-function mutations in the CLCN1 gene. Clinically, they are characterized by myotonia, defined as delayed muscle relaxation after voluntary contraction, which leads to symptoms of muscle stiffness, pain, fatigue, and weakness. Diagnosis is based on history and examination findings, the presence of electrical myotonia on electromyography, and genetic confirmation. In the absence of genetic confirmation, the diagnosis is supported by detailed electrophysiological testing, exclusion of other related disorders, and analysis of a variant of uncertain significance if present. Symptomatic treatment with a sodium channel blocker, such as mexiletine, is usually the first step in management, as well as educating patients about potential anesthetic complications.
Keywords: management; myotonia congenita; nondystrophic myotonias; paramyotonia congenita; skeletal muscle channelopathies.
© 2020 Wiley Periodicals, Inc.
Figures
References
-
- Koch MC, Steinmeyer K, Lorenz C, et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992; 257:797–800. - PubMed
-
- McClatchey AI, Van den Bergh P, Pericak-Vance MA, et al. Temperature-sensitive mutations in the III-IV cytoplasmic loop region of the skeletal muscle sodium channel gene in paramyotonia congenita. Cell. 1992;68:769–774. - PubMed
-
- Strümpell A Tonische Krämpfe in willkürliche bewegten Muskeln (Myotonia congenita). Berl Klin Wochenchr. 1881;9:119–121.
-
- Erb W Klinischen und Pathologisch-Anatomisches von der Thomsenschen Krankheit. Neurol Centralblb. 1885;13:289–194.
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
