

Copy number variation in human genomes from three major ethno-linguistic groups in Africa
- PMID: 32272904
- PMCID: PMC7147055
- DOI: 10.1186/s12864-020-6669-y
Copy number variation in human genomes from three major ethno-linguistic groups in Africa
Abstract
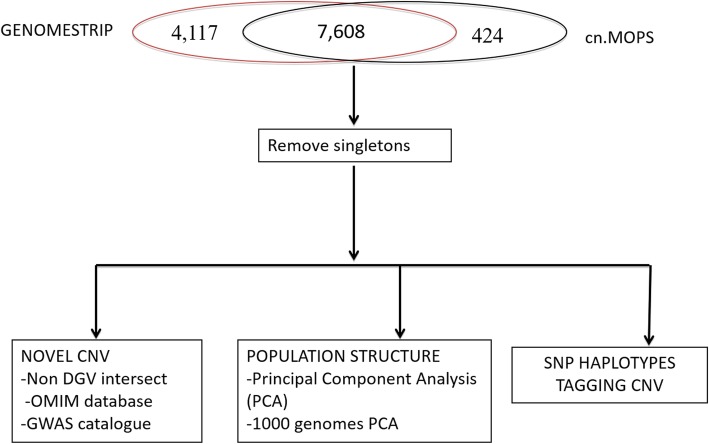
Background: Copy number variation is an important class of genomic variation that has been reported in 75% of the human genome. However, it is underreported in African populations. Copy number variants (CNVs) could have important impacts on disease susceptibility and environmental adaptation. To describe CNVs and their possible impacts in Africans, we sequenced genomes of 232 individuals from three major African ethno-linguistic groups: (1) Niger Congo A from Guinea and Côte d'Ivoire, (2) Niger Congo B from Uganda and the Democratic Republic of Congo and (3) Nilo-Saharans from Uganda. We used GenomeSTRiP and cn.MOPS to identify copy number variant regions (CNVRs).
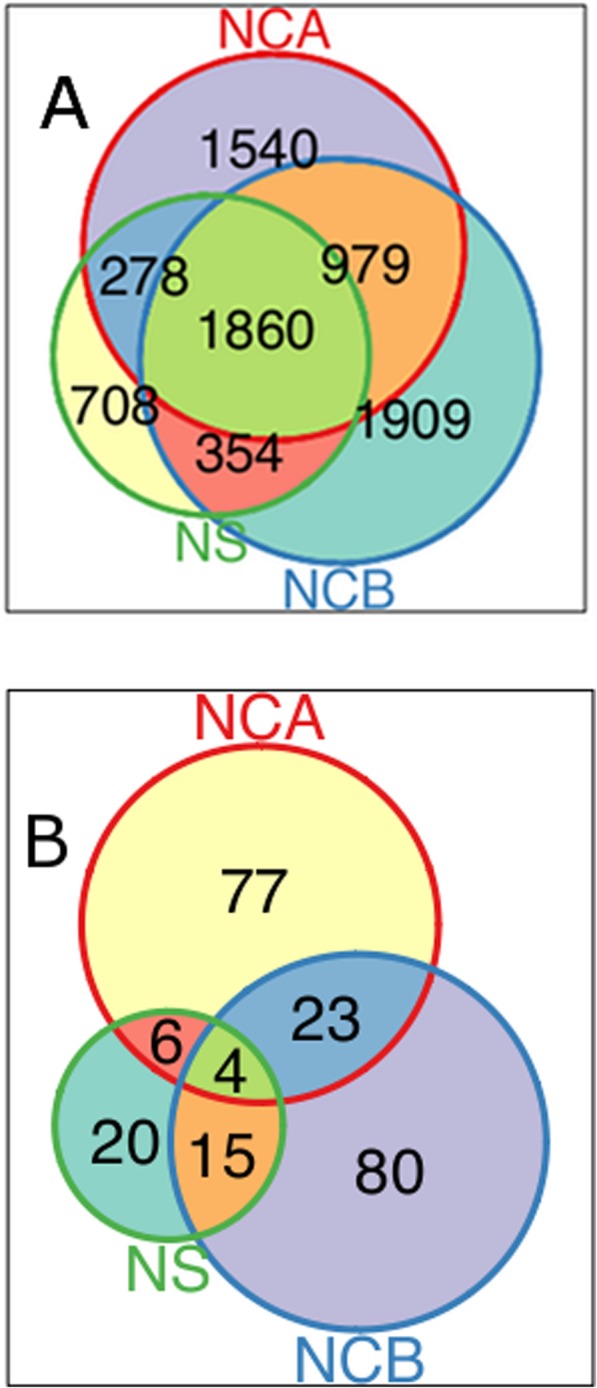
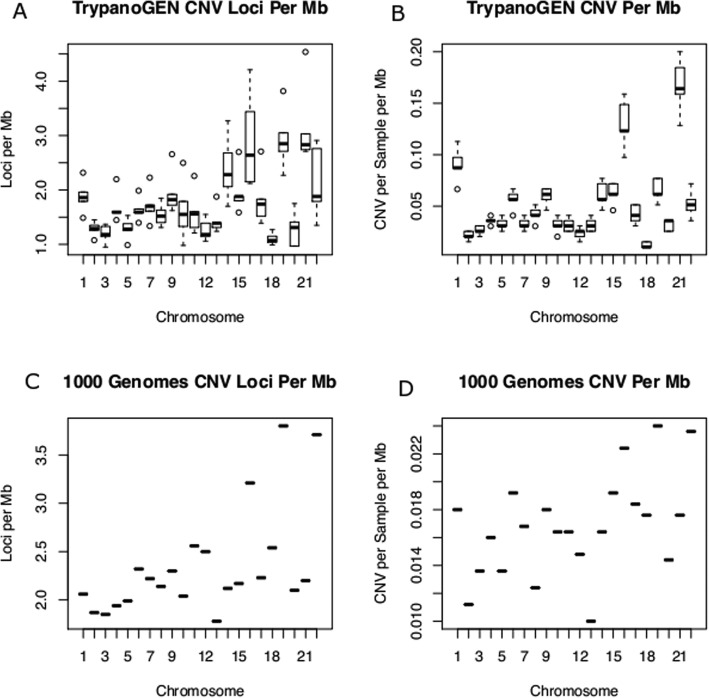
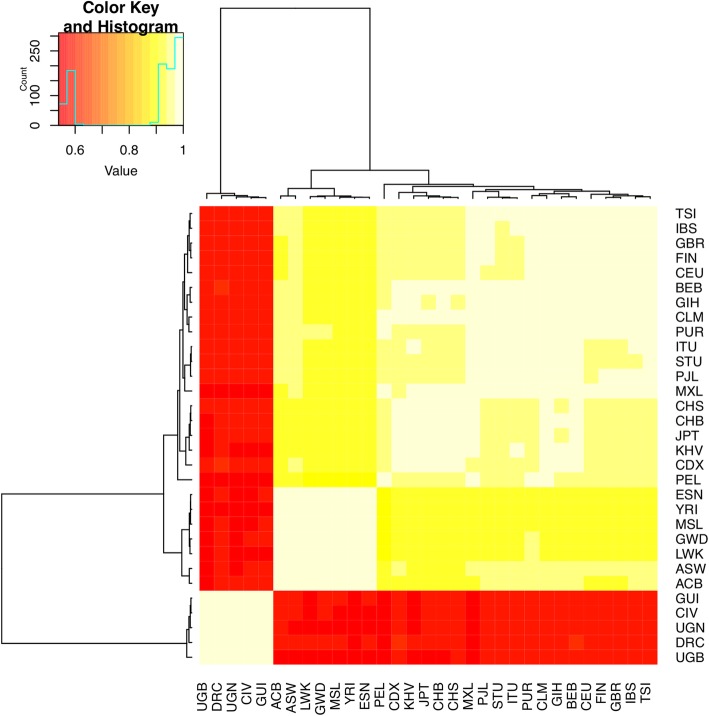
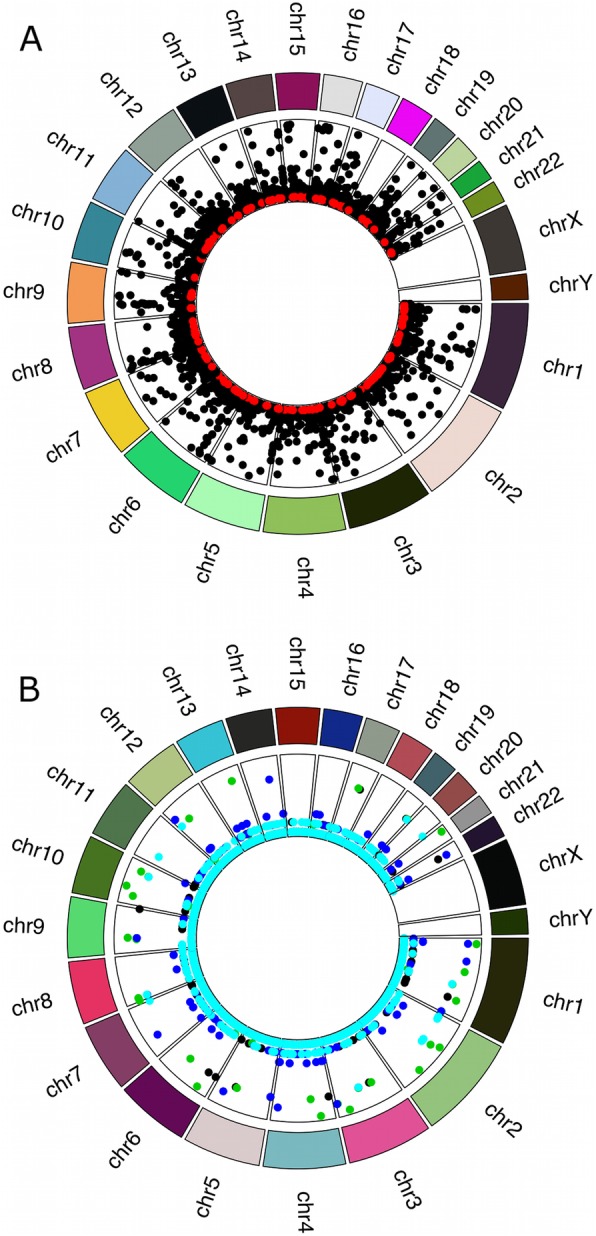
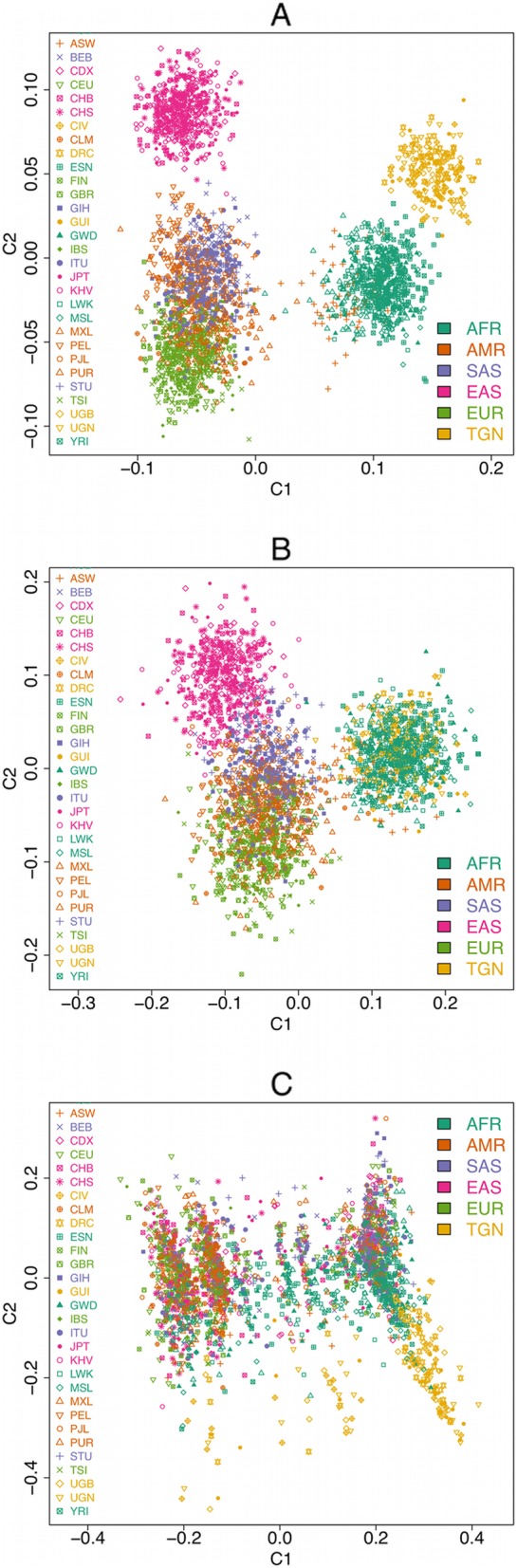
Results: We detected 7608 CNVRs, of which 2172 were only deletions, 2384 were only insertions and 3052 had both. We detected 224 previously un-described CNVRs. The majority of novel CNVRs were present at low frequency and were not shared between populations. We tested for evidence of selection associated with CNVs and also for population structure. Signatures of selection identified previously, using SNPs from the same populations, were overrepresented in CNVRs. When CNVs were tagged with SNP haplotypes to identify SNPs that could predict the presence of CNVs, we identified haplotypes tagging 3096 CNVRs, 372 CNVRs had SNPs with evidence of selection (iHS > 3) and 222 CNVRs had both. This was more than expected (p < 0.0001) and included loci where CNVs have previously been associated with HIV, Rhesus D and preeclampsia. When integrated with 1000 Genomes CNV data, we replicated their observation of population stratification by continent but no clustering by populations within Africa, despite inclusion of Nilo-Saharans and Niger-Congo populations within our dataset.
Conclusions: Novel CNVRs in the current study increase representation of African diversity in the database of genomic variants. Over-representation of CNVRs in SNP signatures of selection and an excess of SNPs that both tag CNVs and are subject to selection show that CNVs may be the actual targets of selection at some loci. However, unlike SNPs, CNVs alone do not resolve African ethno-linguistic groups. Tag haplotypes for CNVs identified may be useful in predicting African CNVs in future studies where only SNP data is available.
Keywords: Adaptation; CNV; Niger Congo A; Niger Congo B; Nilo-Saharan; Signatures of selection; Structural variation; Tag haplotypes.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
