

Synthesis, pharmacological evaluation and structure-activity relationship of recently discovered enzyme antagonist azoles
- PMID: 32274429
- PMCID: PMC7132078
- DOI: 10.1016/j.heliyon.2020.e03656
Synthesis, pharmacological evaluation and structure-activity relationship of recently discovered enzyme antagonist azoles
Abstract
Global people are suffering from the legion of diseases. Cytotoxic property of the chemical compound would not solely influence effective drug properties and reduce unnecessary side effects. Proteins/enzymes responsible for microbe proliferation or survival are specifically targeted and inhibited successfully making the cells to undergo apoptosis. Furthermore, isoforms of essential enzymes have distinct physiological functions; thereby inhibition of essential enzyme isoforms is an apt way to the clinical approach of disease neutralization. Drugs are designed so as to play significant roles such as signaling pathways in the oncogenic process including cell proliferation, invasion, and angiogenesis. The present review comprises collective information of the recent synthesis of various organic drug compounds in brief, which could inhibit particular enzyme. The review also covers the correlation of the structure of a drug molecule designed and its inhibitory activity. Also, the most significant enzyme inhibitors are highlighted and structural moieties/core units responsible for remarkable inhibitory values are emphasized.
Keywords: COX; Carbonic anhydrase; Enzyme inhibitors; Natural product chemistry; Organic chemistry; Pyrazole; Thiazole.
© 2020 Published by Elsevier Ltd.
Figures




























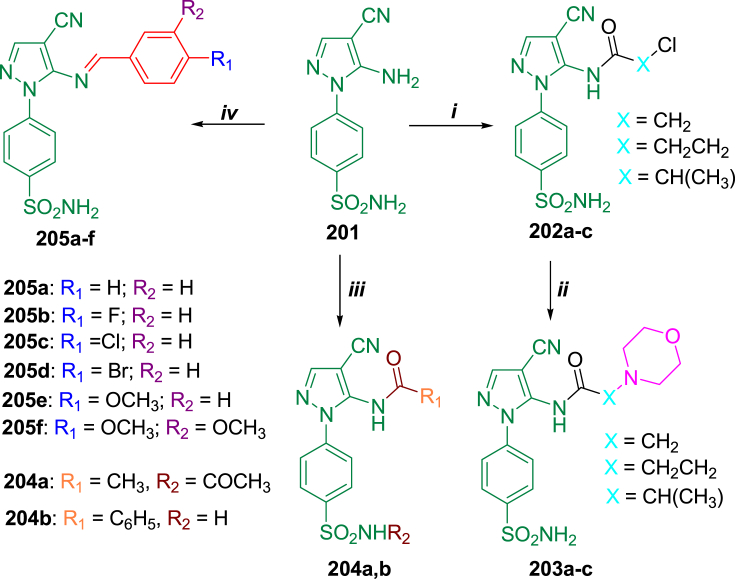
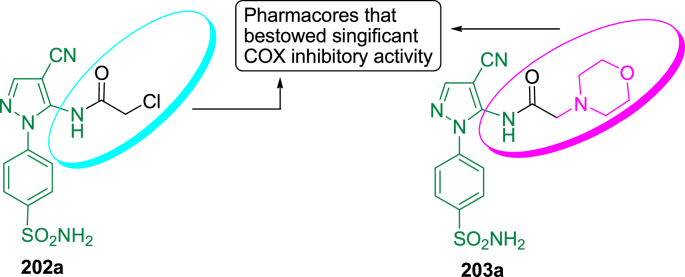

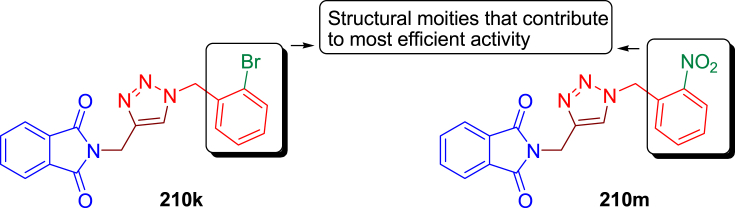
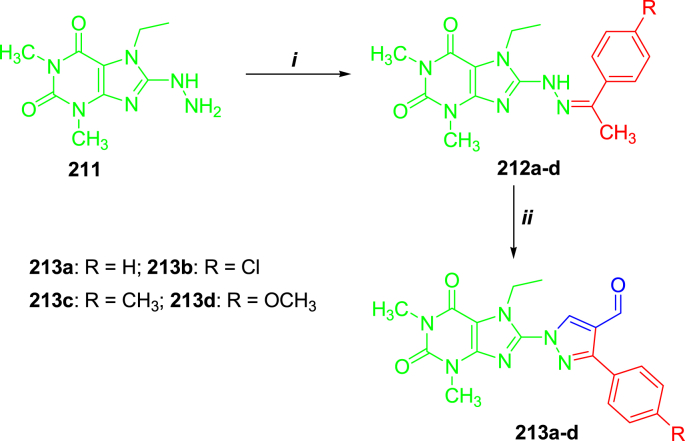








































Similar articles
-
Synthesis, biological evaluation and molecular docking of novel pyrazole derivatives as potent carbonic anhydrase and acetylcholinesterase inhibitors.Bioorg Chem. 2019 May;86:420-427. doi: 10.1016/j.bioorg.2019.02.013. Epub 2019 Feb 5. Bioorg Chem. 2019. PMID: 30769267
-
Synthesis and biological evaluation of novel pyrazoline-based aromatic sulfamates with potent carbonic anhydrase isoforms II, IV and IX inhibitory efficacy.Bioorg Chem. 2018 Apr;77:633-639. doi: 10.1016/j.bioorg.2018.02.021. Epub 2018 Feb 23. Bioorg Chem. 2018. PMID: 29502024
-
Structural Insights for Drugs Developed for Phospholipase D Enzymes.Curr Drug Discov Technol. 2018;15(2):81-93. doi: 10.2174/1570163814666170816112135. Curr Drug Discov Technol. 2018. PMID: 28814238 Review.
-
Isatin-pyrazole benzenesulfonamide hybrids potently inhibit tumor-associated carbonic anhydrase isoforms IX and XII.Eur J Med Chem. 2015 Oct 20;103:583-93. doi: 10.1016/j.ejmech.2015.09.021. Epub 2015 Sep 16. Eur J Med Chem. 2015. PMID: 26408817
-
Multicomponent chemistry in the synthesis of carbonic anhydrase inhibitors.J Enzyme Inhib Med Chem. 2016;31(sup4):185-199. doi: 10.1080/14756366.2016.1220944. Epub 2016 Oct 26. J Enzyme Inhib Med Chem. 2016. PMID: 27784162 Review.
Cited by
-
Design, synthesis, biological evaluation and molecular docking study of novel pleuromutilin derivatives containing substituted benzoxazole as antibacterial agents.J Enzyme Inhib Med Chem. 2023 Dec;38(1):2251712. doi: 10.1080/14756366.2023.2251712. J Enzyme Inhib Med Chem. 2023. PMID: 37664987 Free PMC article.
-
Current knowledge of leptin in wound healing: A collaborative review.Front Pharmacol. 2022 Sep 12;13:968142. doi: 10.3389/fphar.2022.968142. eCollection 2022. Front Pharmacol. 2022. PMID: 36172174 Free PMC article. Review.
References
-
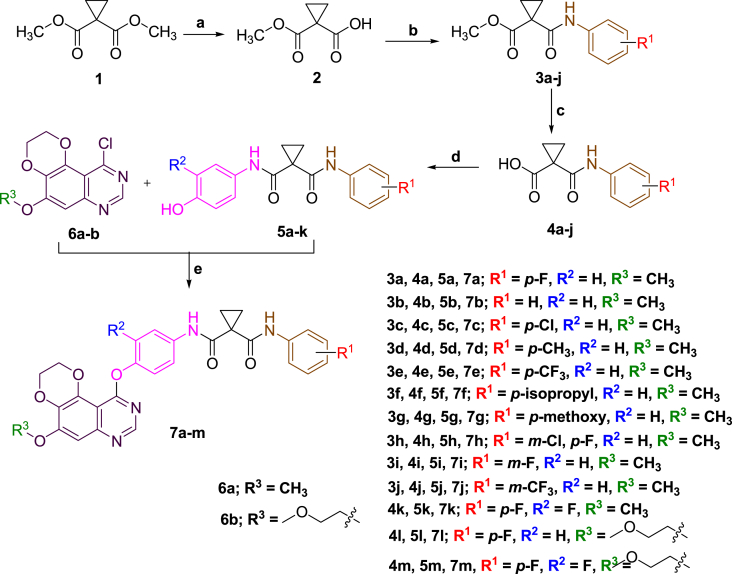
- Wei D., Fan H., n Zheng K., Qin X., Yang L., Yang Y., e Duan Y., Zhang Q., Zeng C., Hu L. Synthesis and anti-tumor activity of [1,4] dioxino [2,3-f] quinazoline derivatives as dual inhibitors of c-met and VEGFR-2. Bioorg. Inside Chem. 2019;88:10291. - PubMed
-
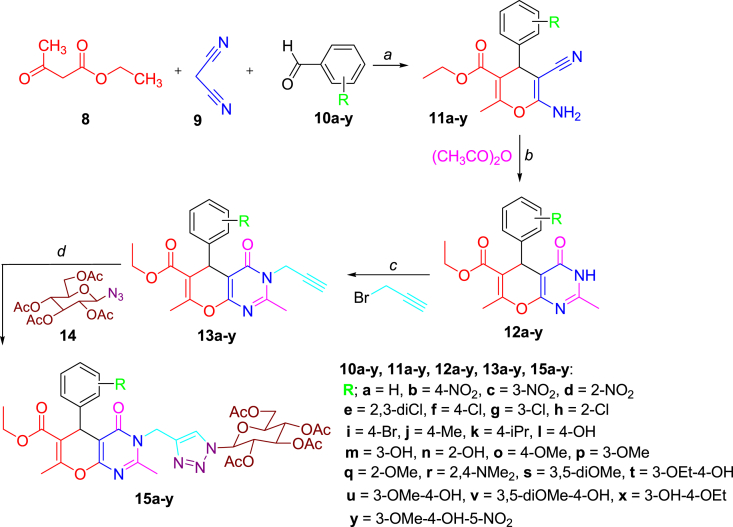
- Thanh N.D., Hai D.S., Ha N.T.T., Tung D.T., Le C.T., Van H.T.K., Toan V.N., Toan D.N., Dang L.H. Synthesis, biological evaluation and molecular docking study of 1,2,3-1Htriazoles having 4H-pyrano[2,3-d]pyrimidine as potential Mycobacterium tuberculosis protein tyrosine phosphatase B inhibitors. Bioorg. Med. Chem. Lett. 2019;29:164–171. - PubMed
-
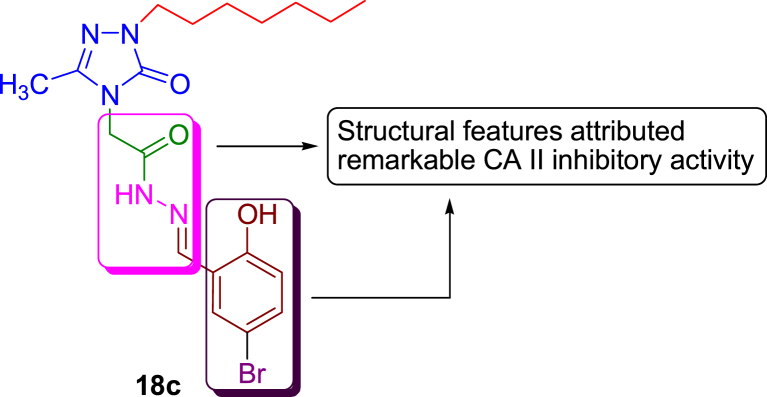
- Akin Safak, Ayaloglu Hasan, Gultekin Ergun, Colak Ahmet, Bekircan Olcay, Yildirim Akatin Melike. Synthesis of 1,2,4-triazole-5-on derivatives and determination of carbonic anhydrase II isoenzyme inhibition effects. Bioorg. Chem. 2019;83:170–179. - PubMed
-
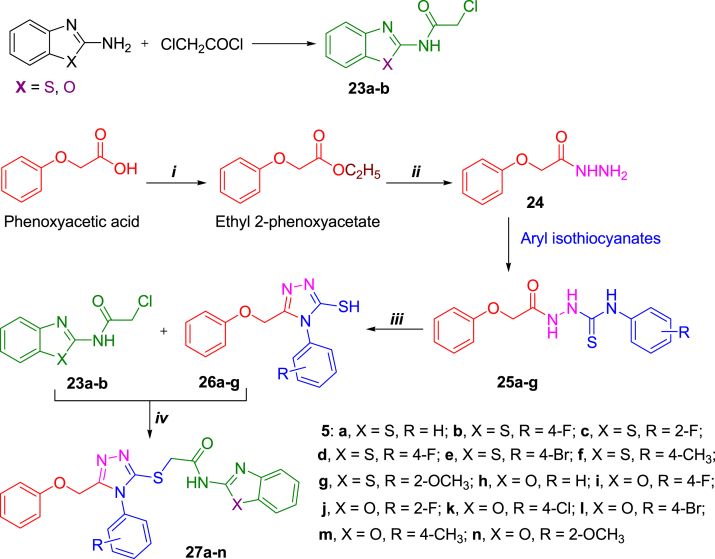
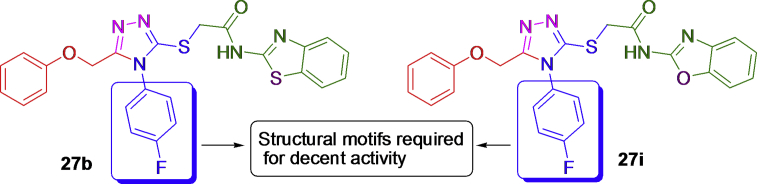
- Tariq S., Kamboj P., Alam O., Amir M. 1,2,4-Triazole-Based benzothiazole/benzoxazole derivatives: design, synthesis, p38α MAP kinase inhibition, anti-inflammatory activity and molecular docking studies. Bioorg. Chem. 2018;81:630–641. - PubMed
-
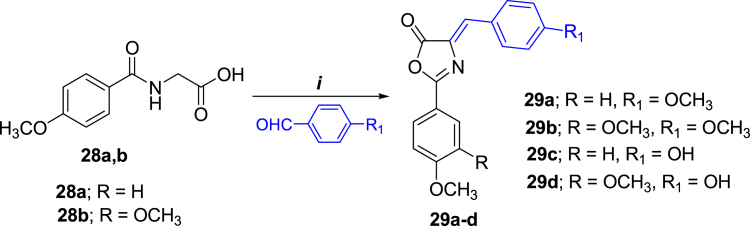
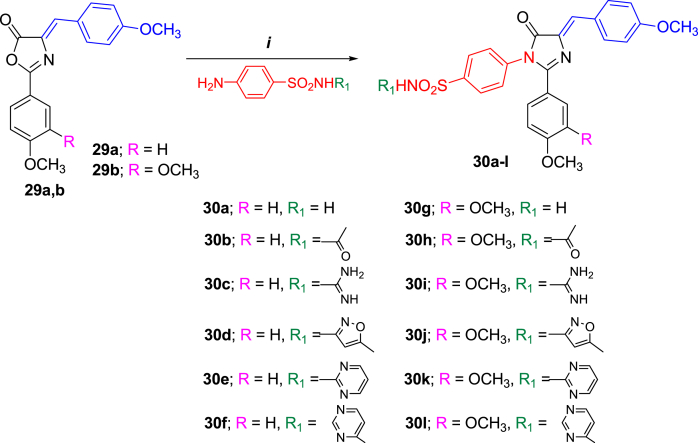
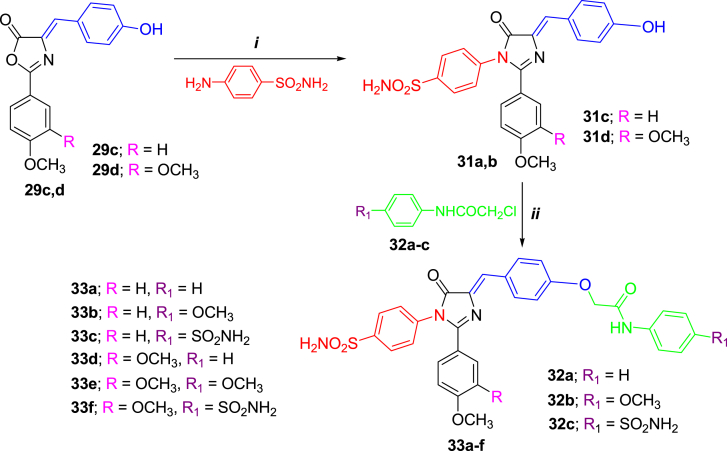
- Georgey H.H., Manhi F.M., Mahmoud W.R., Mohamed N.A., Berrino E., Supuran C.T. 1,2,4-Trisubstituted imidazolinones with dual carbonic anhydrase and p38 mitogen-activated protein kinase inhibitory activity. Bioorg. Chem. 2019;82:109–116. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
