

Development of a neurologic severity scale for Aicardi Goutières Syndrome
- PMID: 32279991
- PMCID: PMC7366613
- DOI: 10.1016/j.ymgme.2020.03.008
Development of a neurologic severity scale for Aicardi Goutières Syndrome
Erratum in
-
Corrigendum to "Development of a neurologic severity scale for Aicardi Goutières Syndrome" [Mol Genet Metab. 2020 Jun;130(2):153-160. PMID: 32279991].Mol Genet Metab. 2022 May;136(1):81. doi: 10.1016/j.ymgme.2022.03.010. Epub 2022 Apr 11. Mol Genet Metab. 2022. PMID: 35422341 No abstract available.
Abstract
Background and purpose: Aicardi Goutières Syndrome (AGS) is a severe, autoinflammatory leukodystrophy characterized by global neurologic dysfunction. Our goal was to create an easy-to-apply scale relevant to the unique developmental challenges associated with AGS.
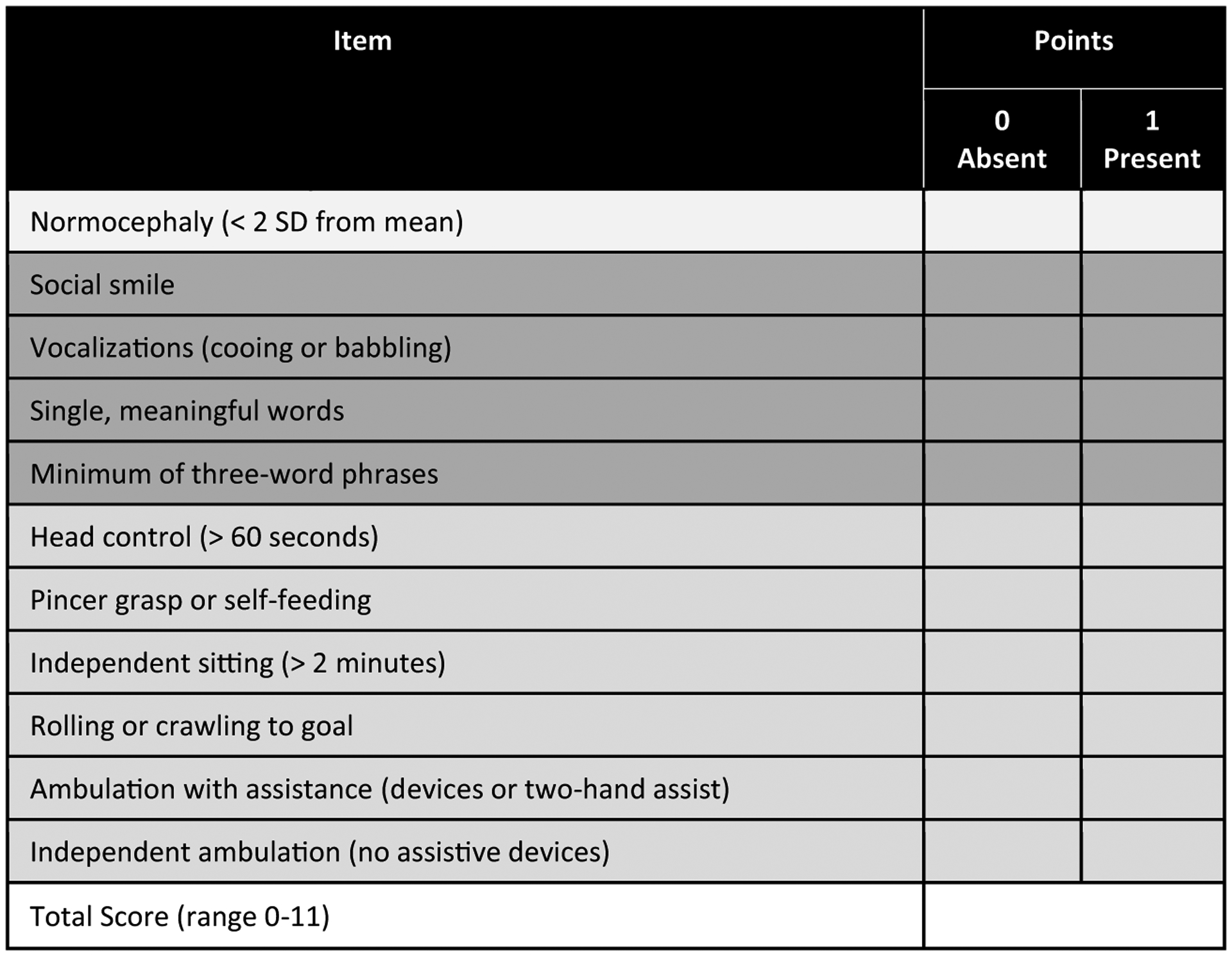
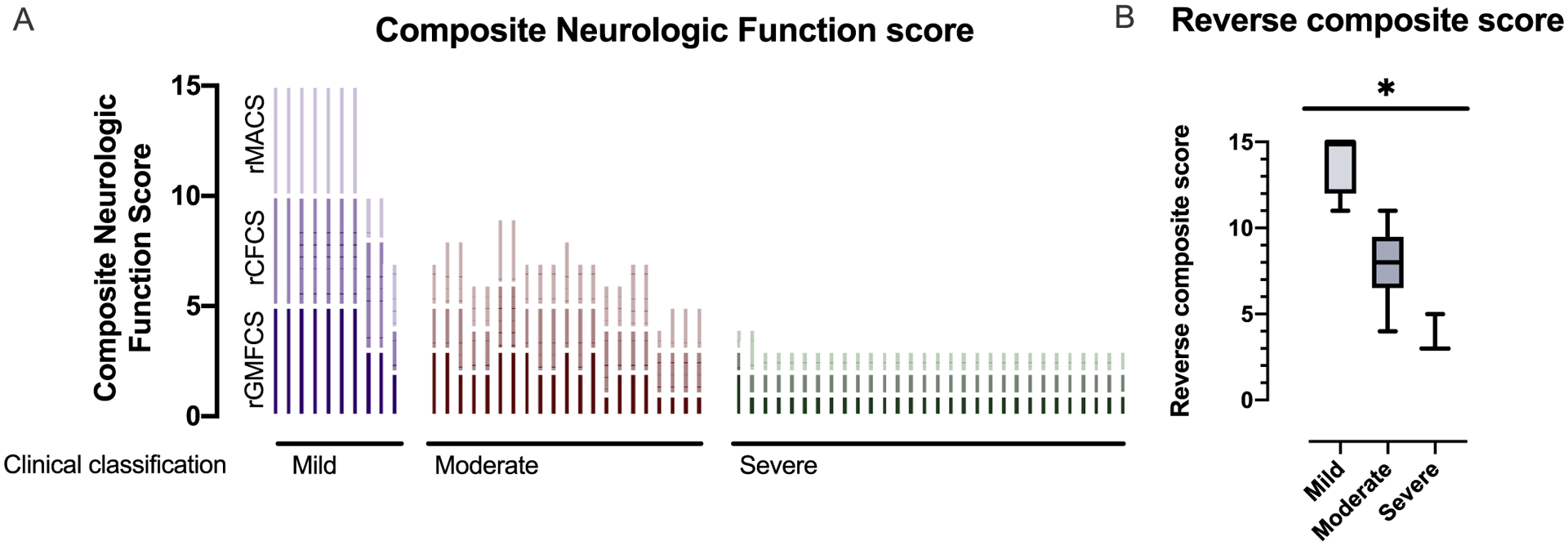
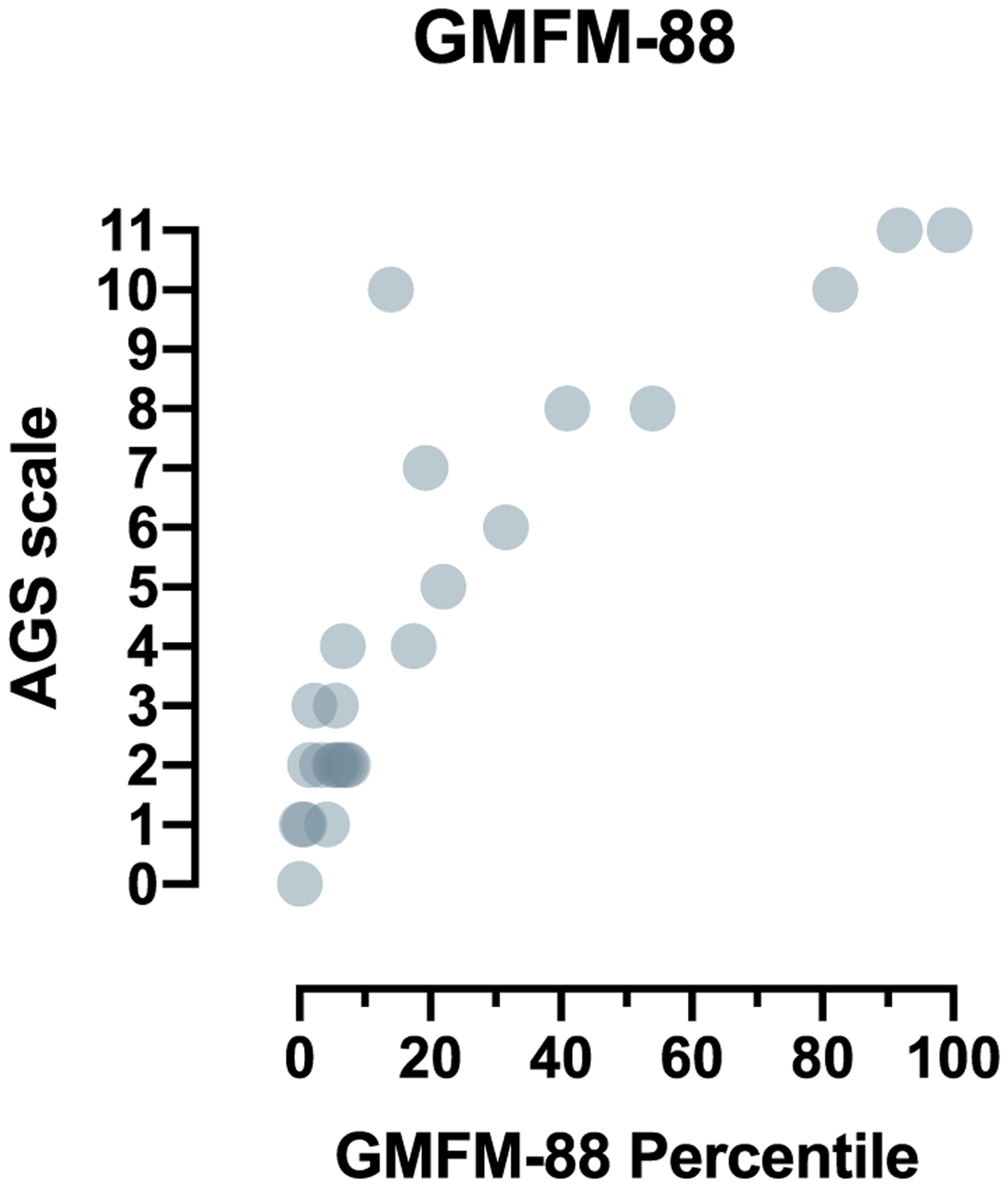
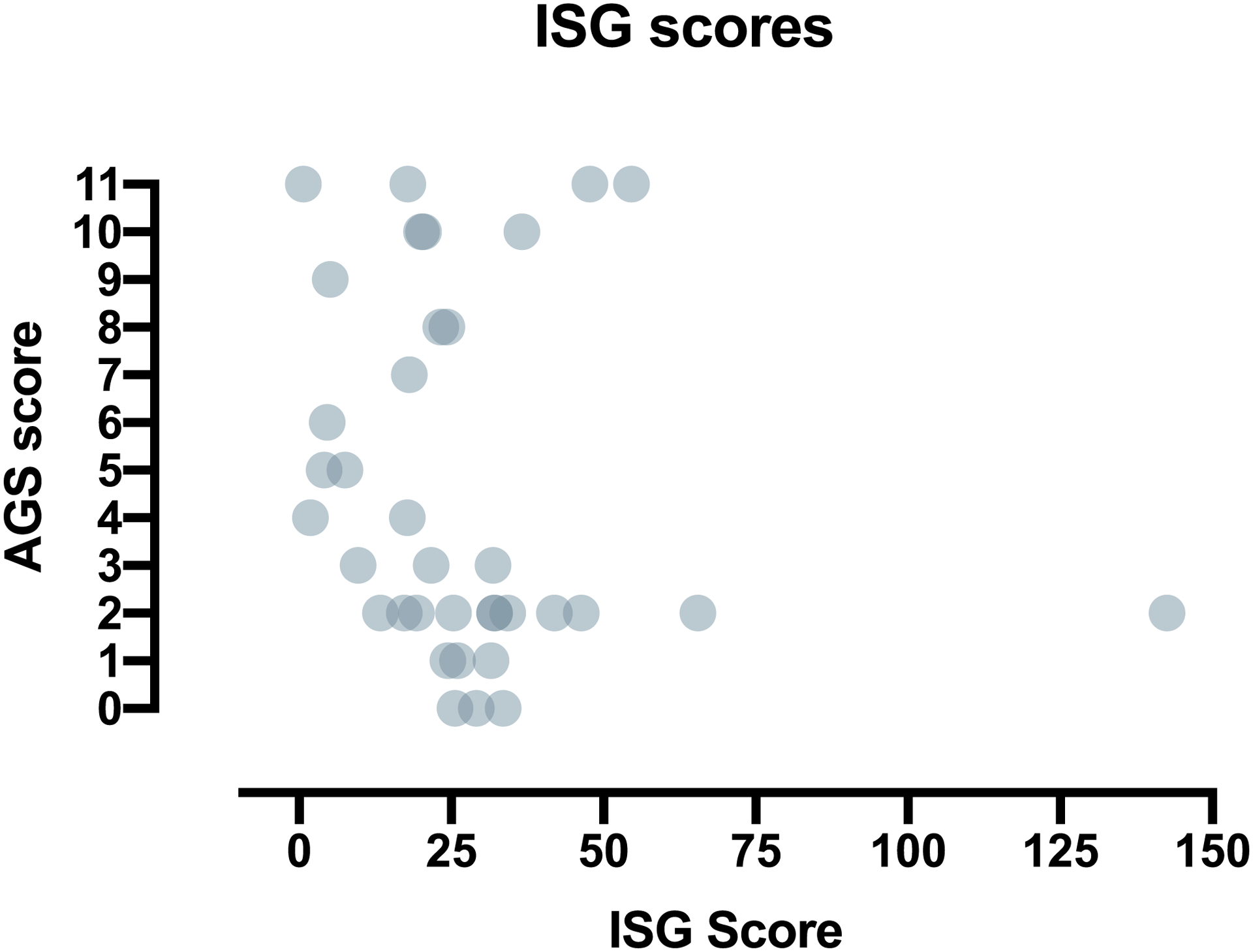
Methods: All individuals were recruited through our natural history study. Individuals were classified by AGS severity as mild, moderate, or severe, and clinical encounters were assigned a composite score for neurologic function calculated from the sum of three functional classification scales. Through expert consensus, we identified 11 key items to reflect the severity of AGS across gross motor, fine motor, and cognitive skills to create the AGS Scale. There was strong interrater reliability. The AGS scale was applied across available medical records to evaluate neurologic function over time. The AGS scale was compared to performance on a standard measure of gross motor function (Gross Motor Function Measure-88, GMFM-88) and a putative diagnostic biomarker of disease, the interferon signaling gene expression score (ISG).
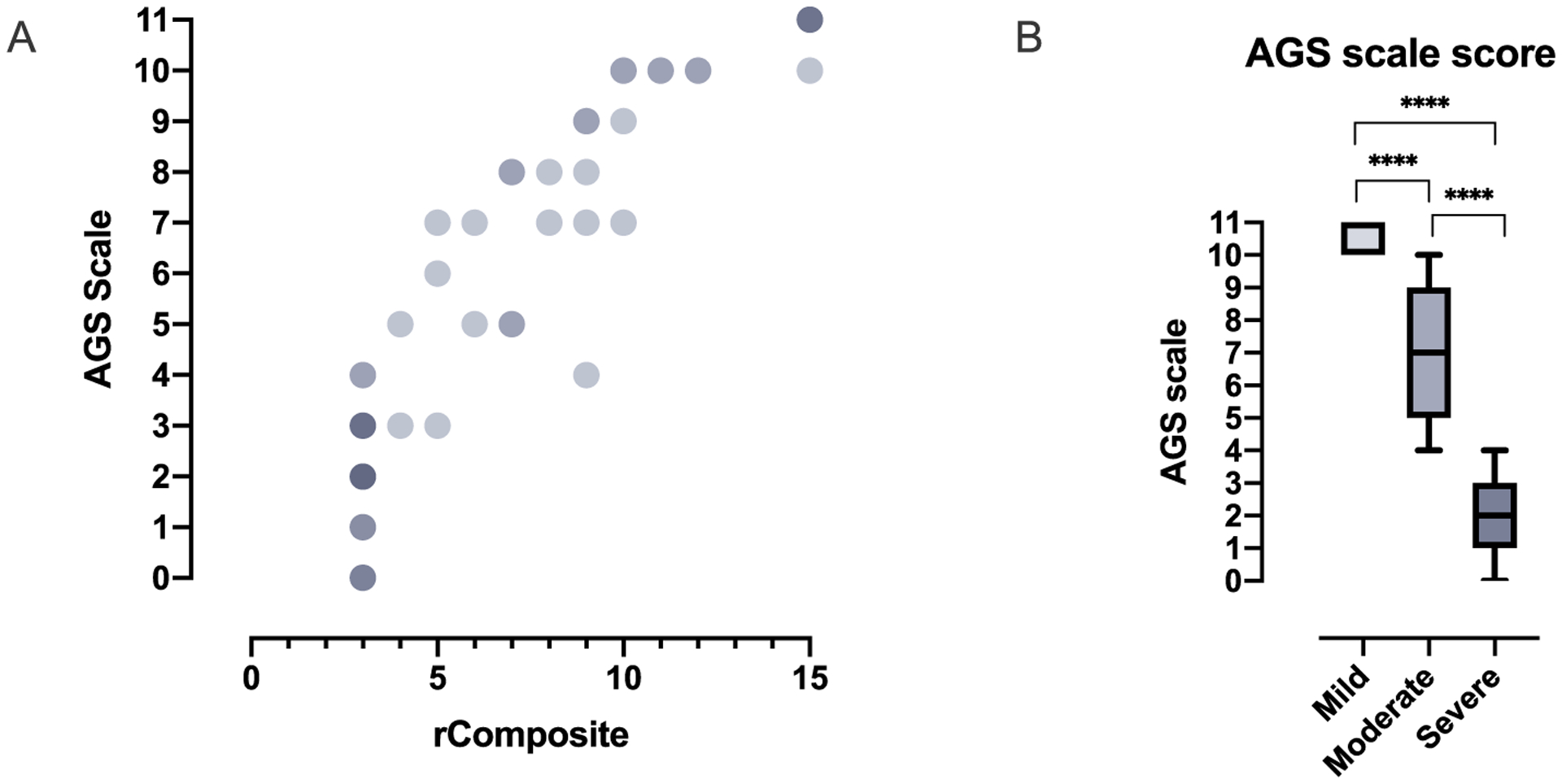
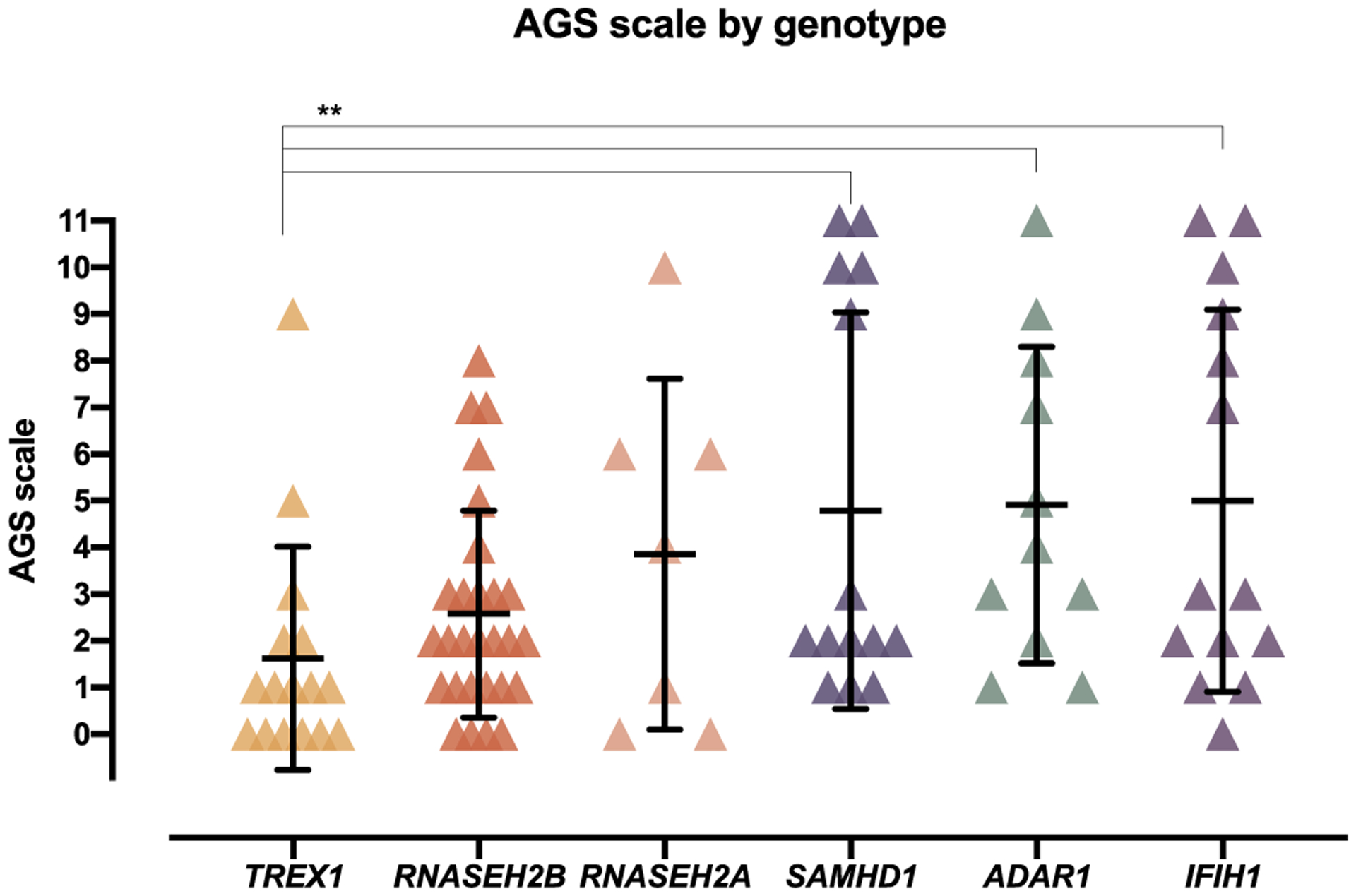
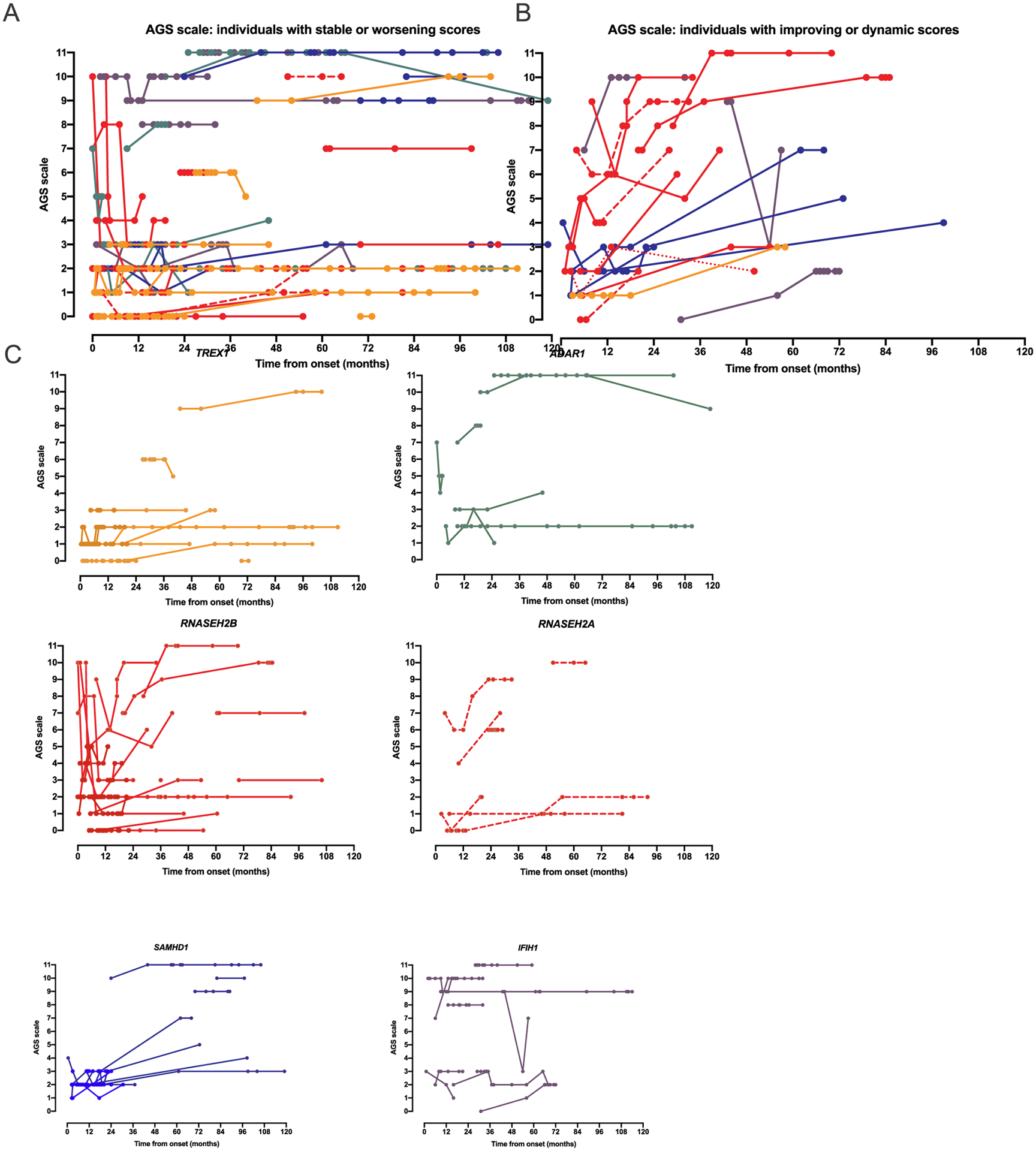
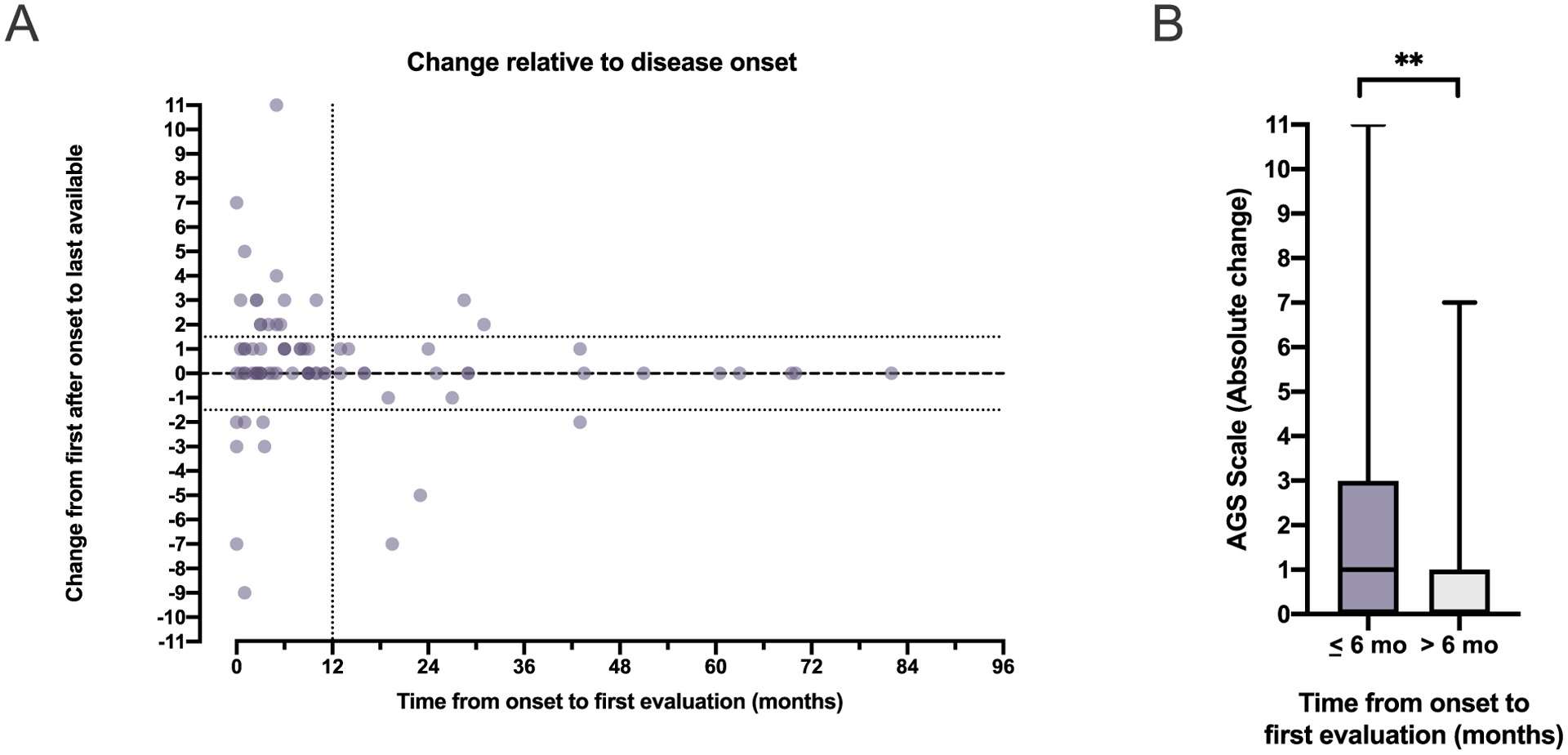
Results: The AGS scale score correlated with severity classifications and the composite neurologic function scores. When retrospectively applied across our natural history study, the majority of individuals demonstrated an initial decline in function followed by stable scores. Within the first 6 months of disease, the AGS score was the most dynamic. The AGS scale correlated with performance by the GMFM-88, but did not correlate with ISG levels.
Conclusions: This study demonstrates the utility of the AGS scale as a multimodal tool for the assessment of neurologic function in AGS. The AGS scale correlates with clinical severity and with a more labor-intensive tool, GMFM-88. This study underscores the limitations of the ISG score as a marker of disease severity. With the AGS scale, we found that AGS neurologic severity is the most dynamic early in disease. This novel AGS scale is a promising tool to longitudinally follow neurologic function in this unique population.
Keywords: Genetic; Interferonopathy; Leukodystrophy; Neurodegenerative; Outcome measure.
Copyright © 2020. Published by Elsevier Inc.
Figures
References
-
- Armangue T, Orsini JJ, Takanohashi A, Gavazzi F, Conant A, Ulrick N, et al., Neonatal detection of Aicardi Goutieres Syndrome by increased C26:0 lysopho-sphatidylcholine and interferon signature on newborn screening blood spots, Mol. Genet. Metab (2017), 10.1016/j.ymgme.2017.07.006. - DOI - PMC - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
