

Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response
- PMID: 32283785
- PMCID: PMC7230342
- DOI: 10.3390/genes11040409
Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response
Abstract
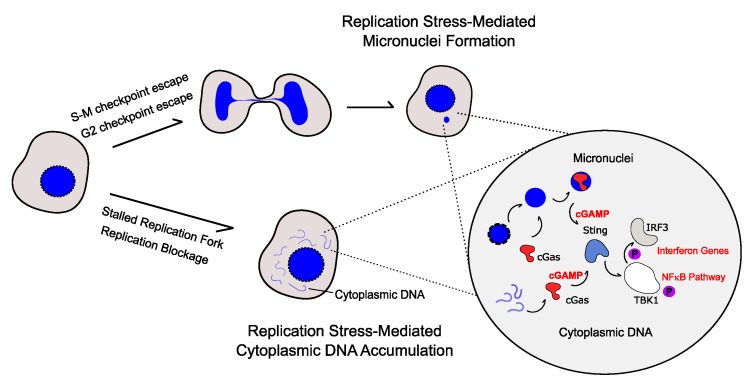
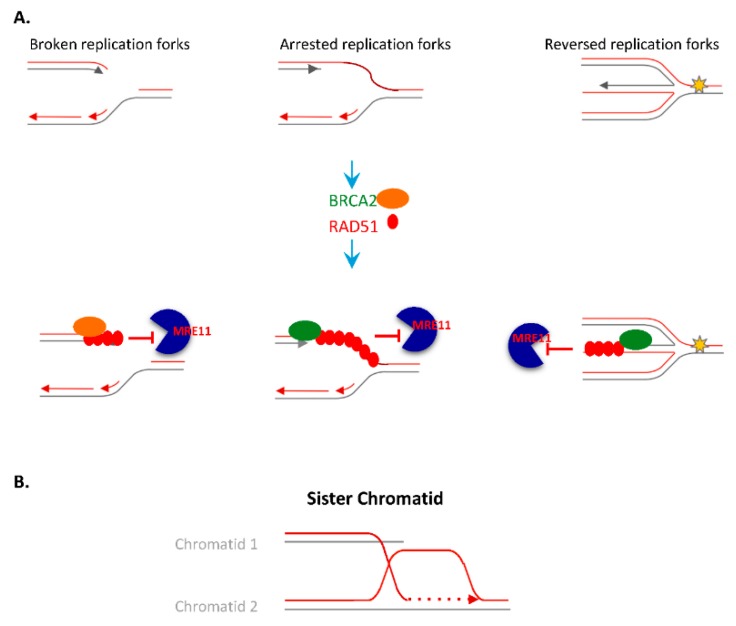
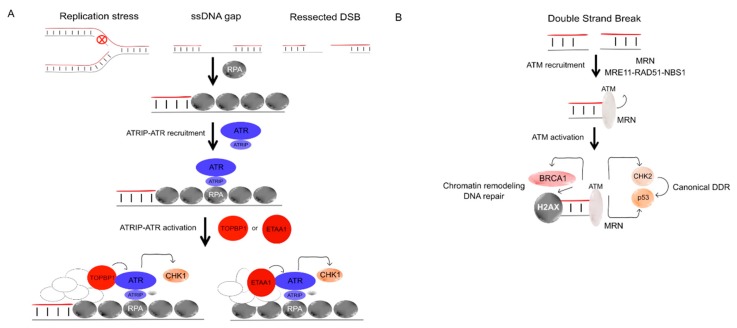
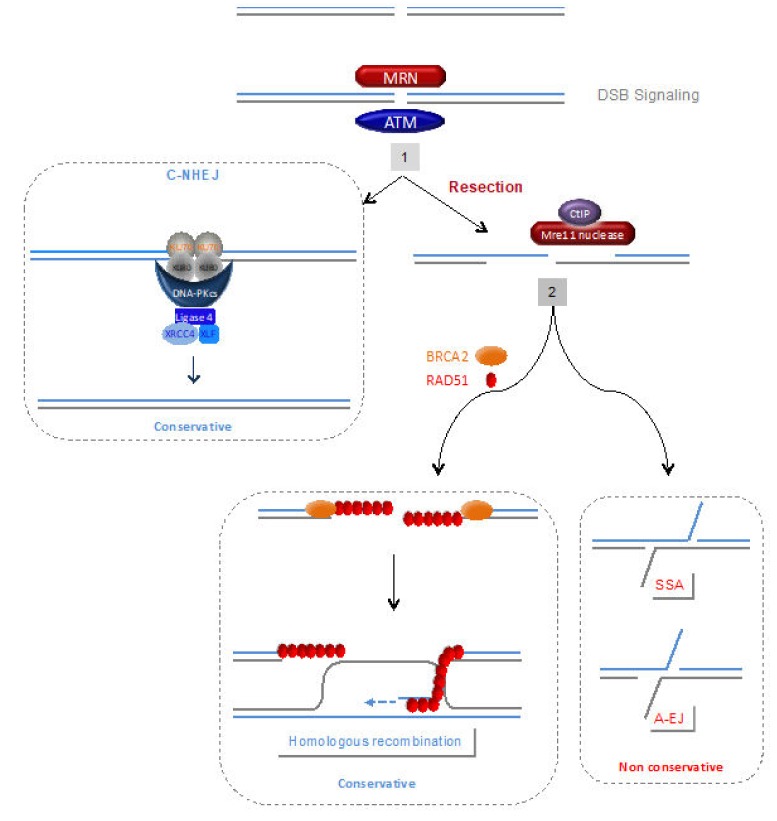
Complete and accurate DNA replication is essential to genome stability maintenance during cellular division. However, cells are routinely challenged by endogenous as well as exogenous agents that threaten DNA stability. DNA breaks and the activation of the DNA damage response (DDR) arising from endogenous replication stress have been observed at pre- or early stages of oncogenesis and senescence. Proper detection and signalling of DNA damage are essential for the autonomous cellular response in which the DDR regulates cell cycle progression and controls the repair machinery. In addition to this autonomous cellular response, replicative stress changes the cellular microenvironment, activating the innate immune response that enables the organism to protect itself against the proliferation of damaged cells. Thereby, the recent descriptions of the mechanisms of the pro-inflammatory response activation after replication stress, DNA damage and DDR defects constitute important conceptual novelties. Here, we review the links of replication, DNA damage and DDR defects to innate immunity activation by pro-inflammatory paracrine effects, highlighting the implications for human syndromes and immunotherapies.
Keywords: DNA damage response; DNA repair; cGAS-STING; inflammation; innate immunity; replicative stress.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
