

Bacterial colonization reprograms the neonatal gut metabolome
- PMID: 32284564
- PMCID: PMC8052915
- DOI: 10.1038/s41564-020-0694-0
Bacterial colonization reprograms the neonatal gut metabolome
Abstract
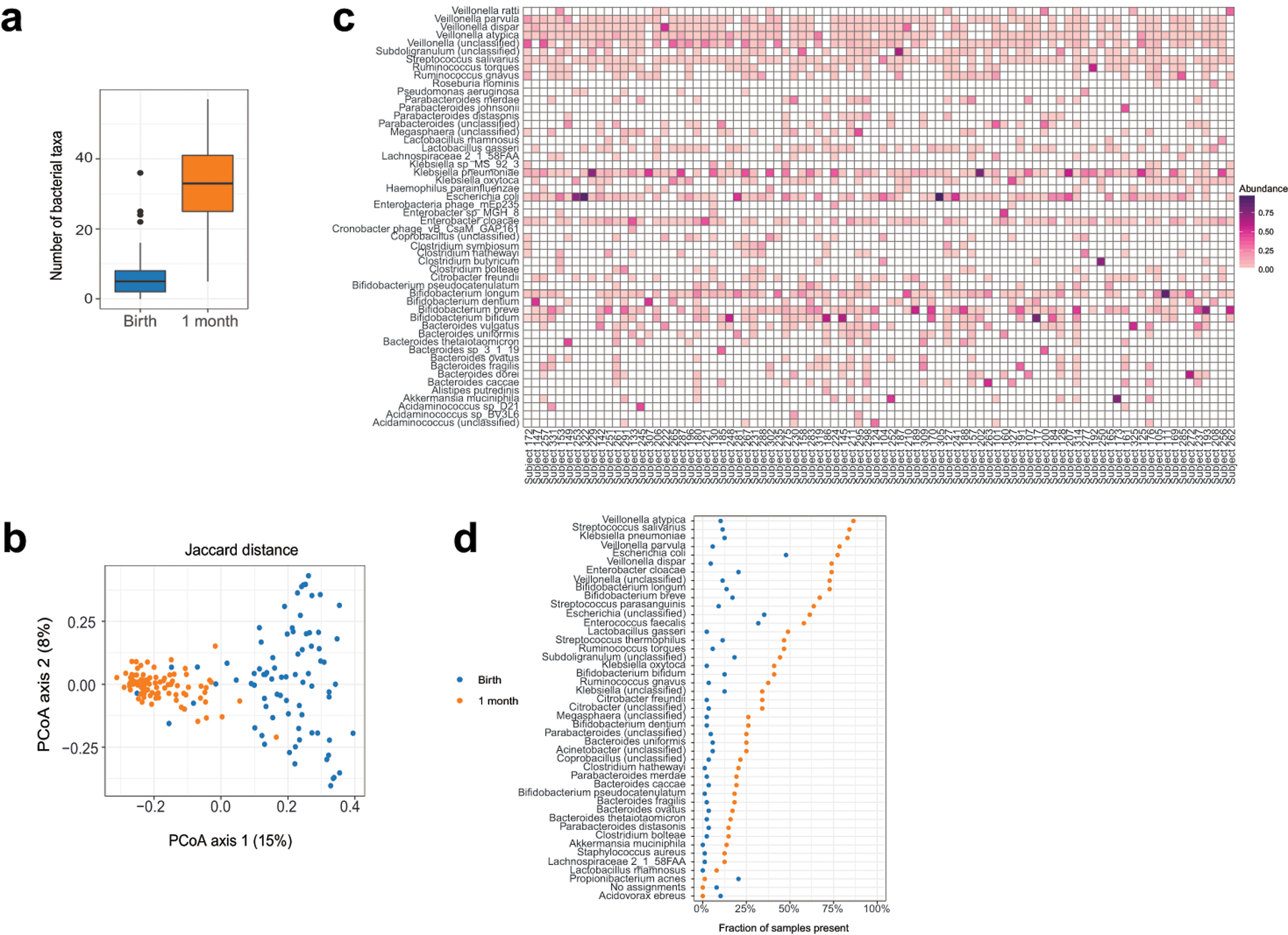
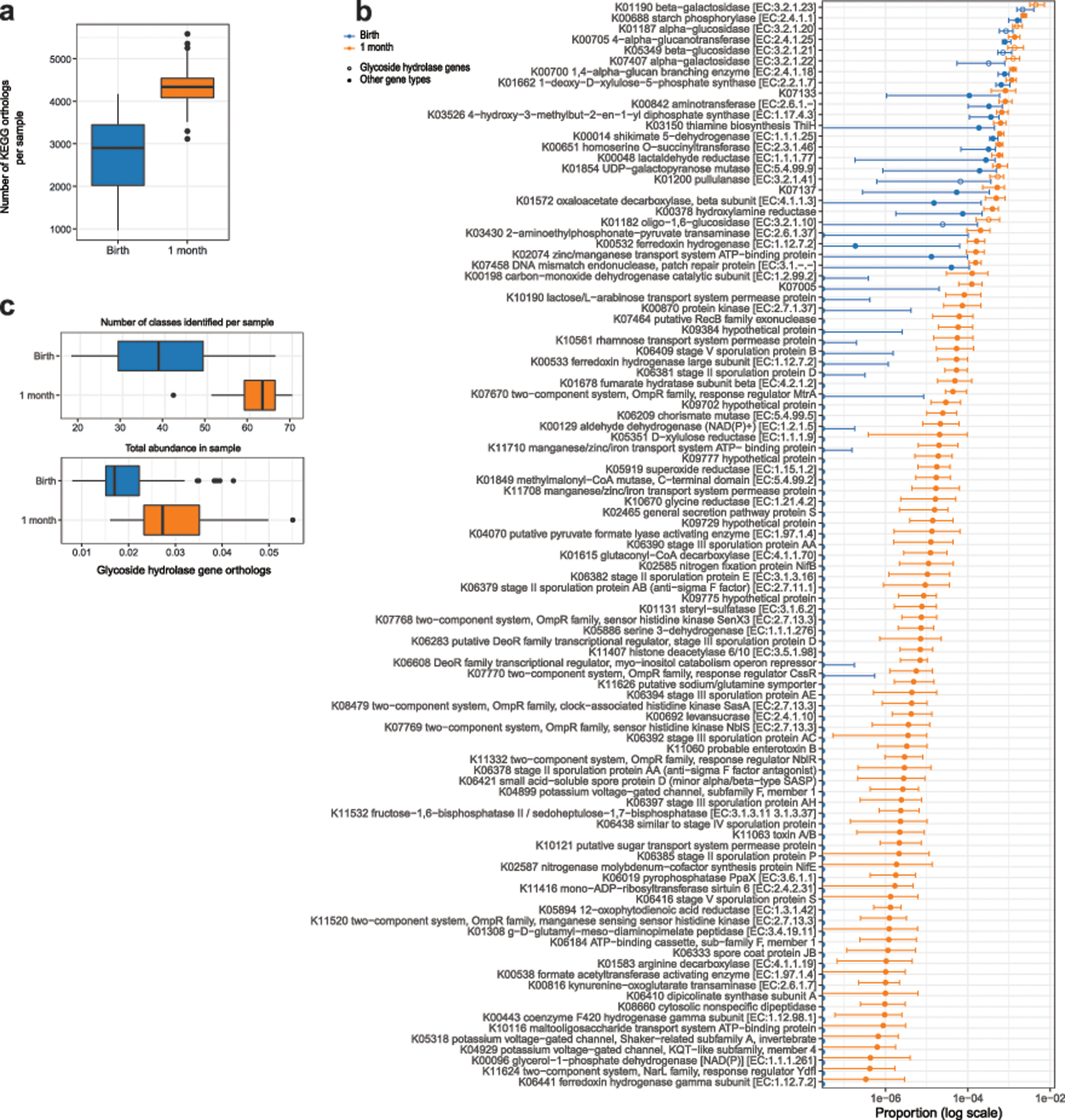
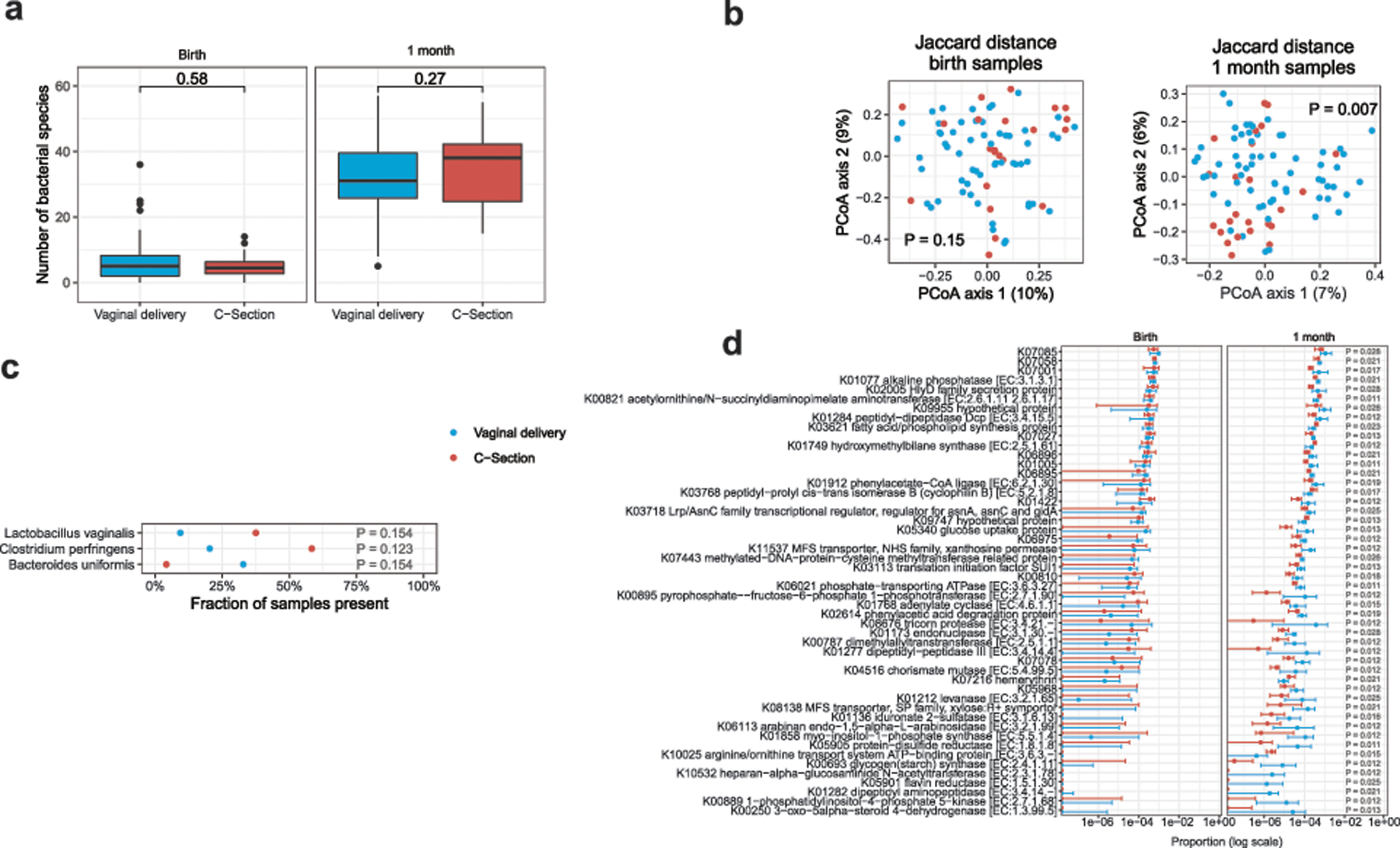
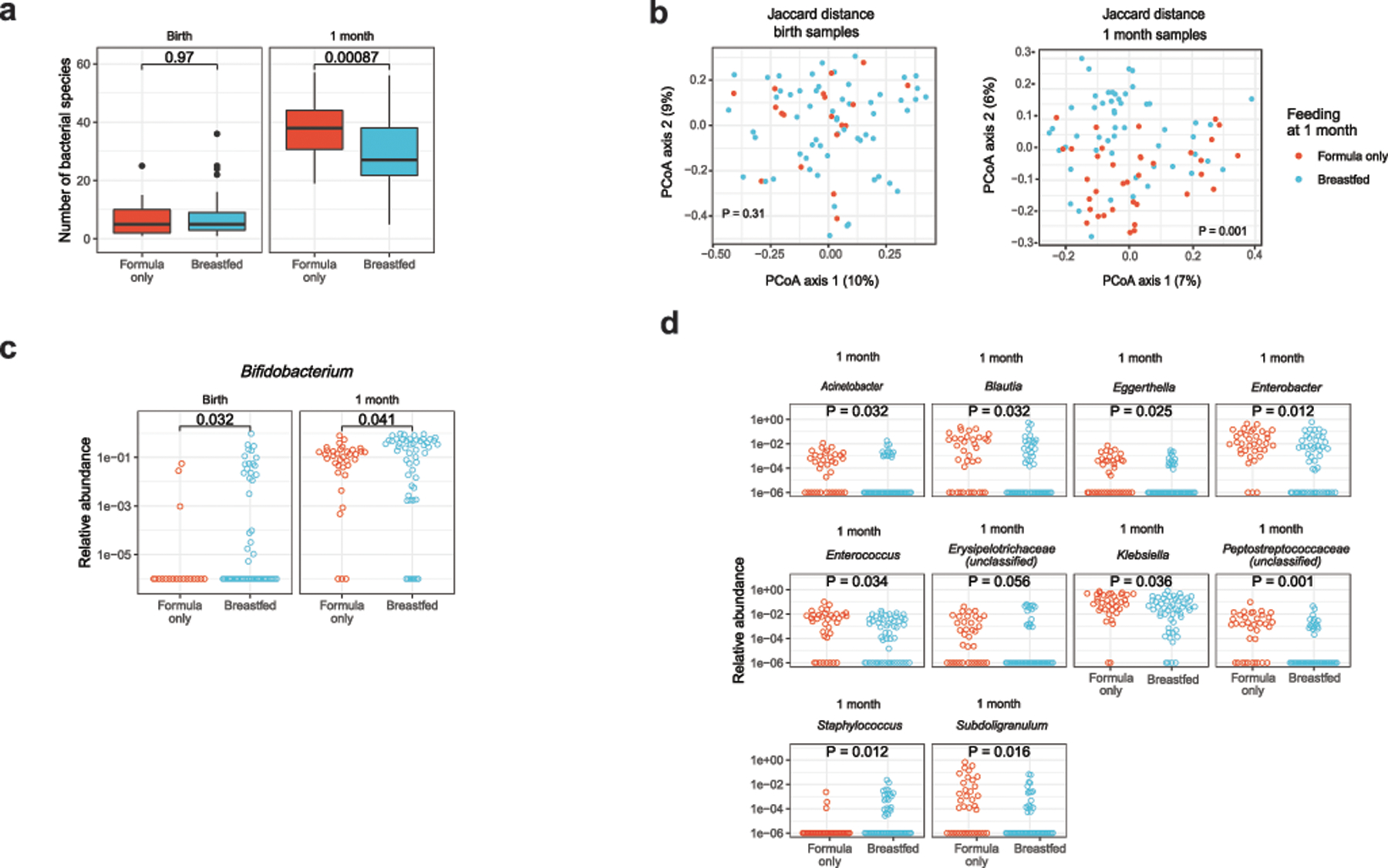
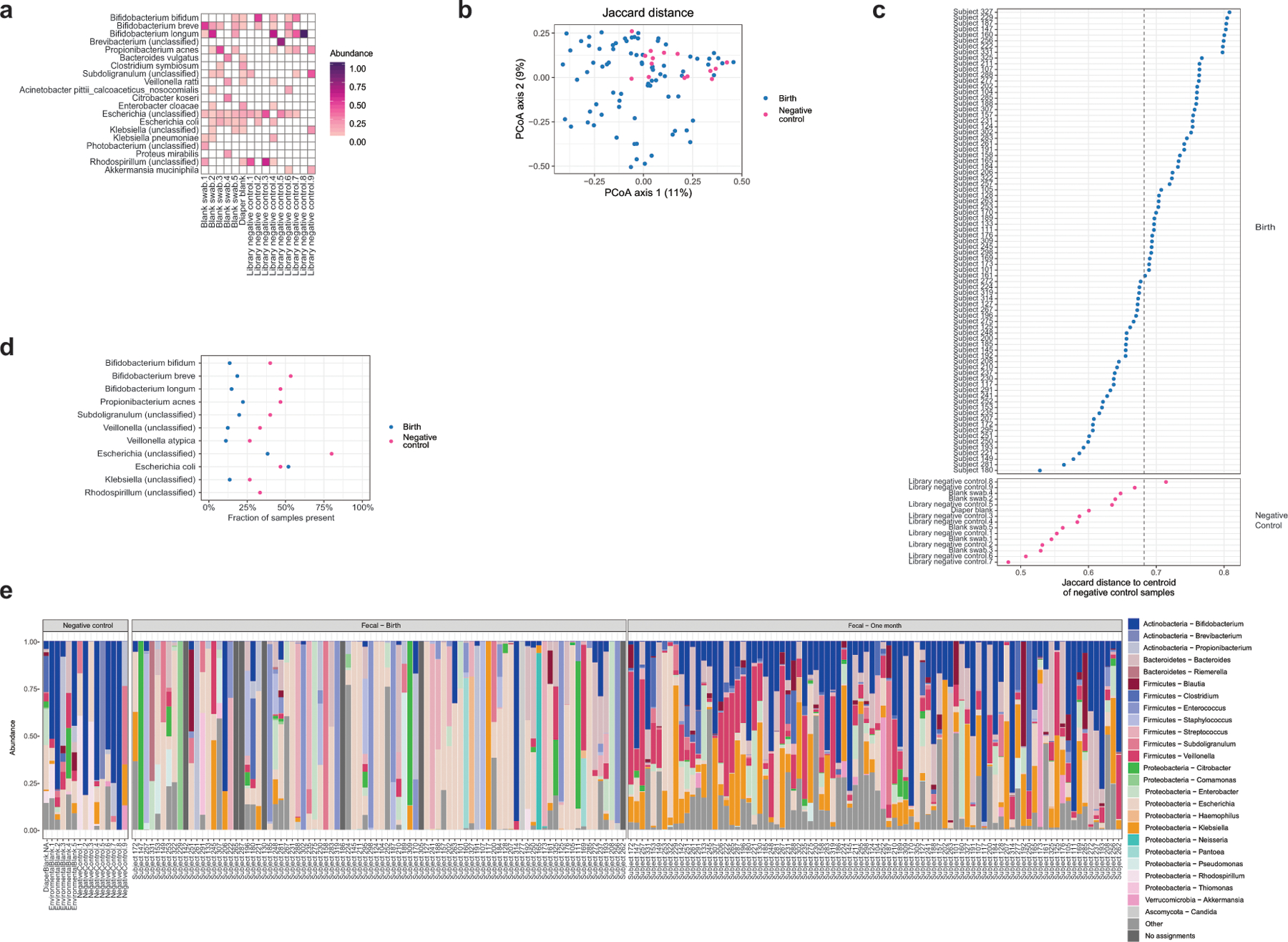
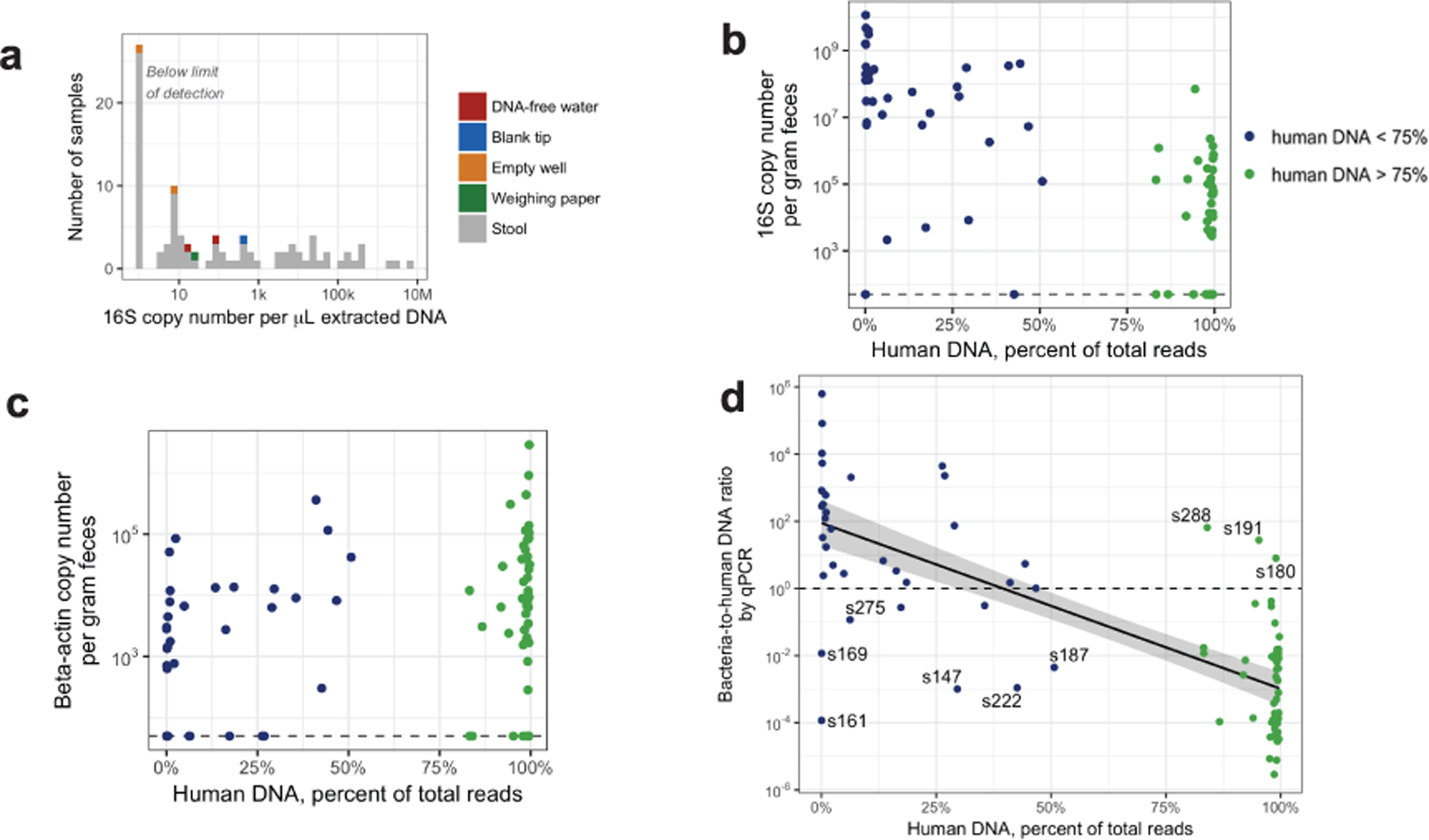
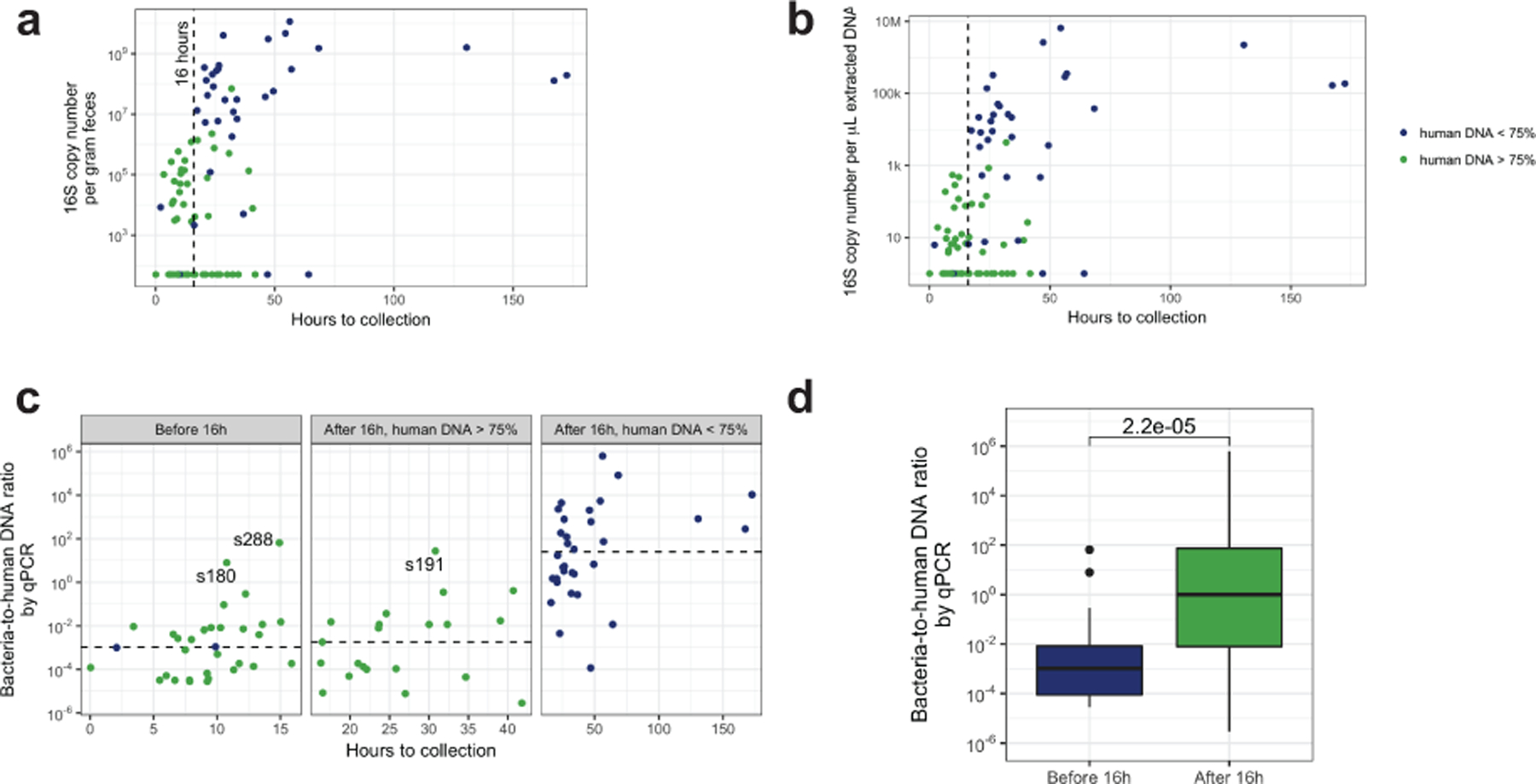
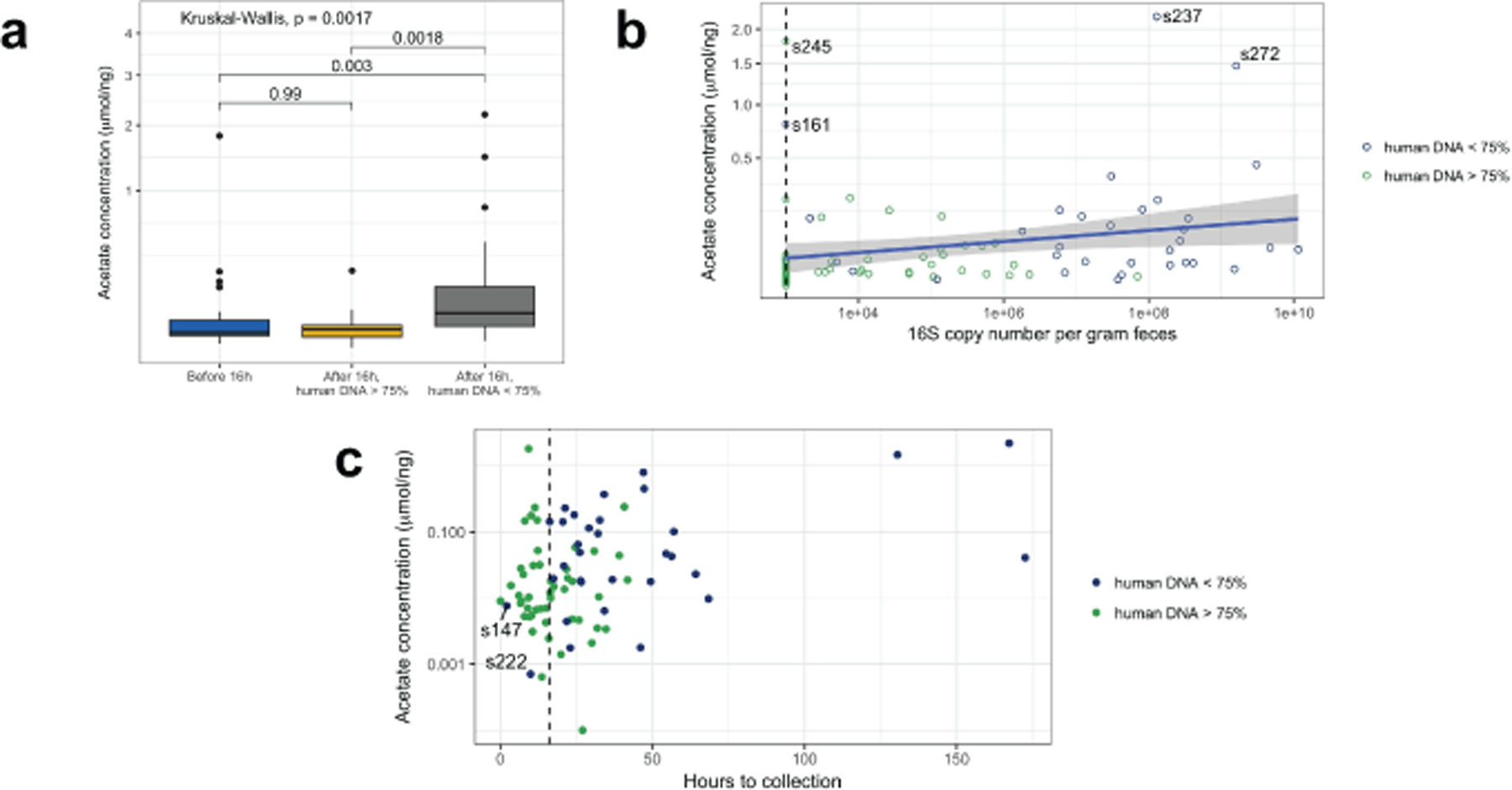
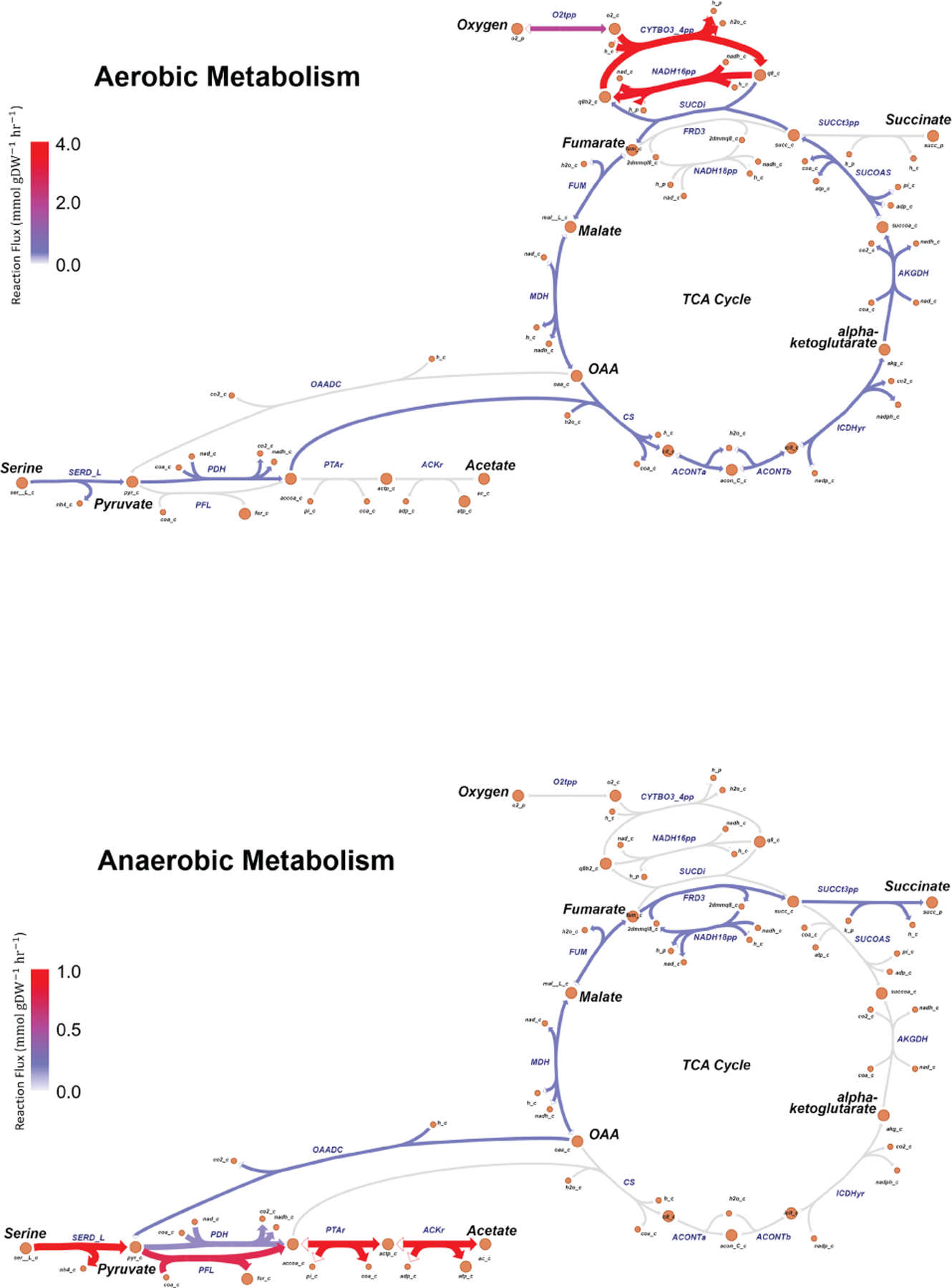
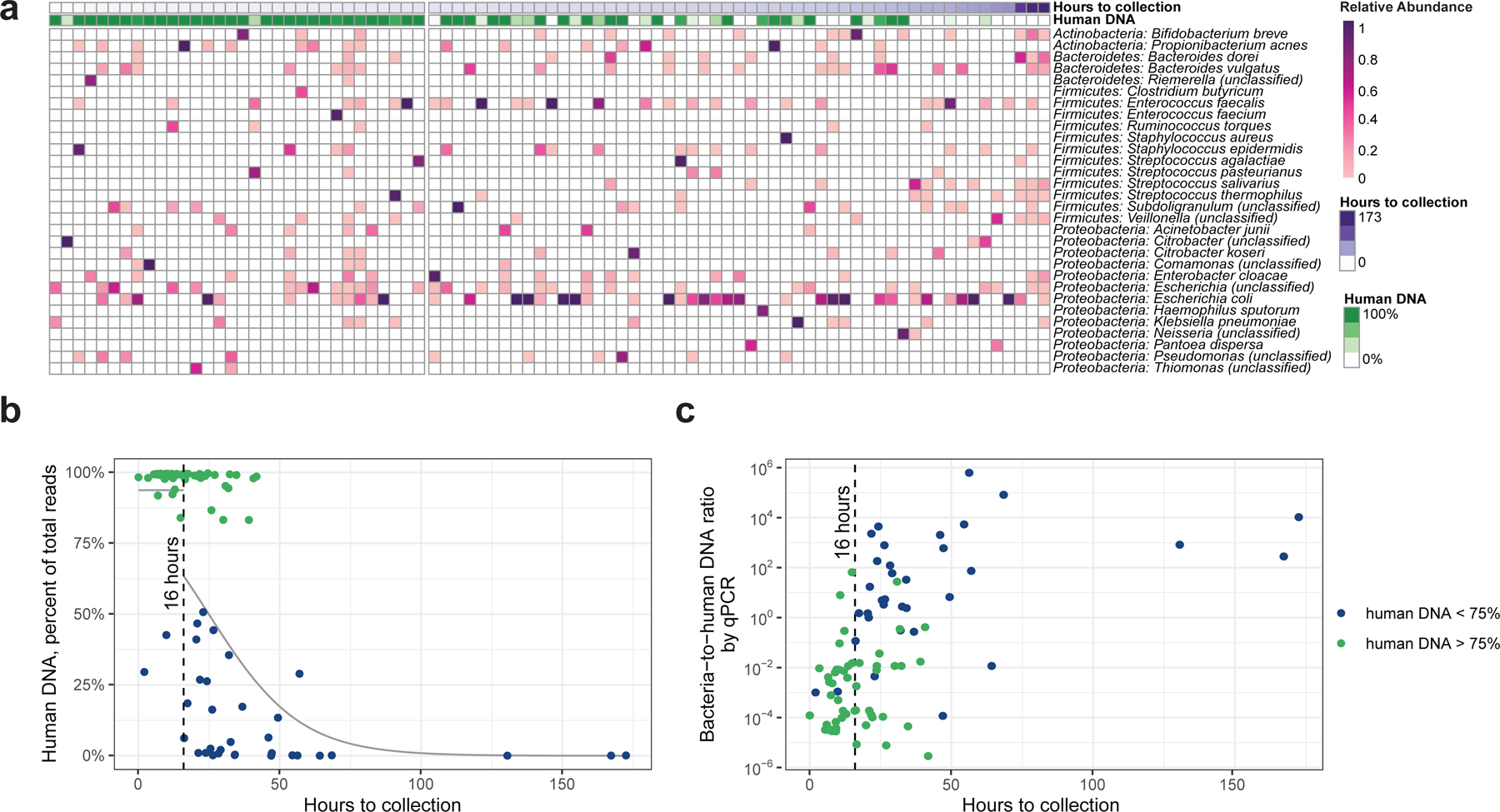
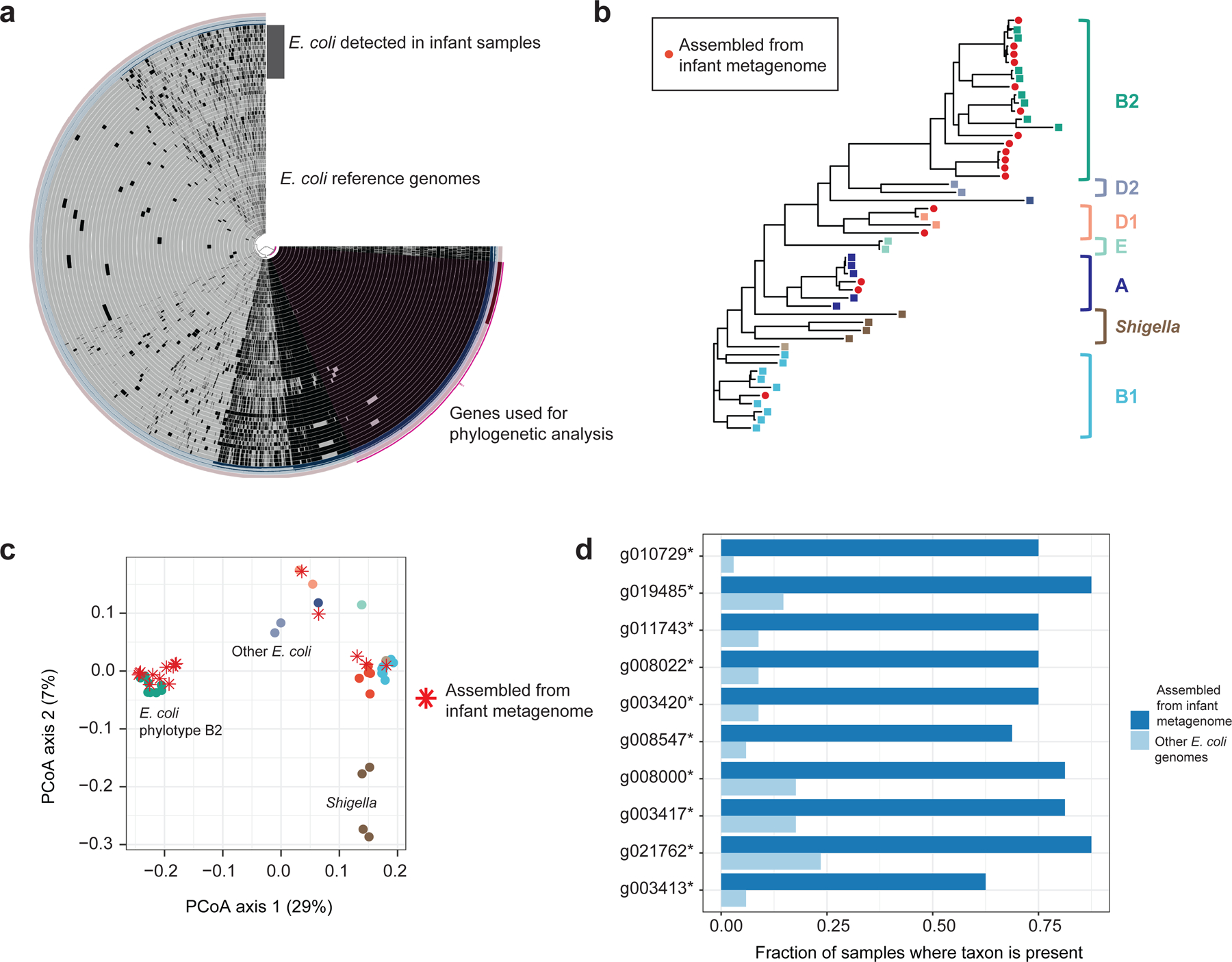
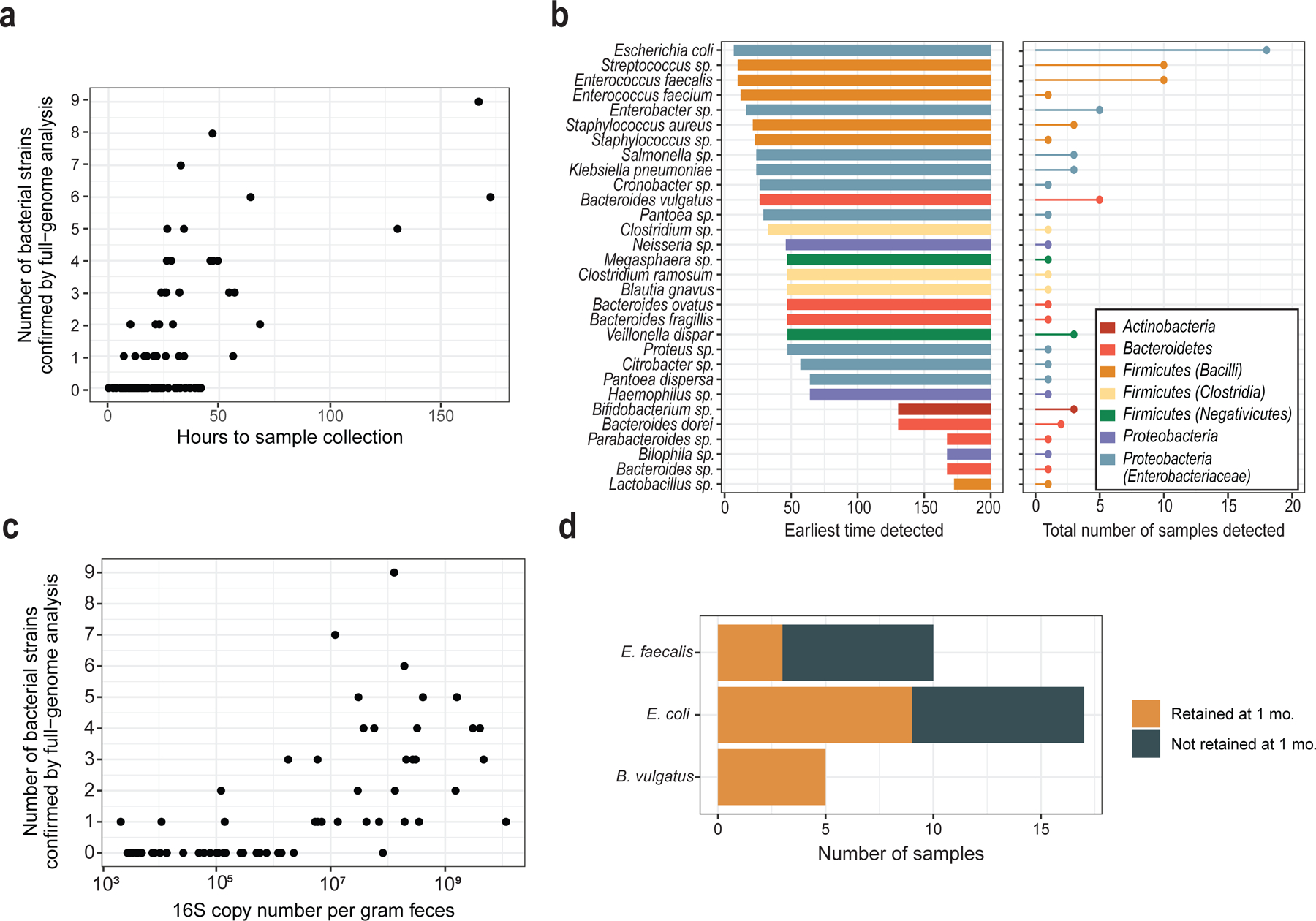
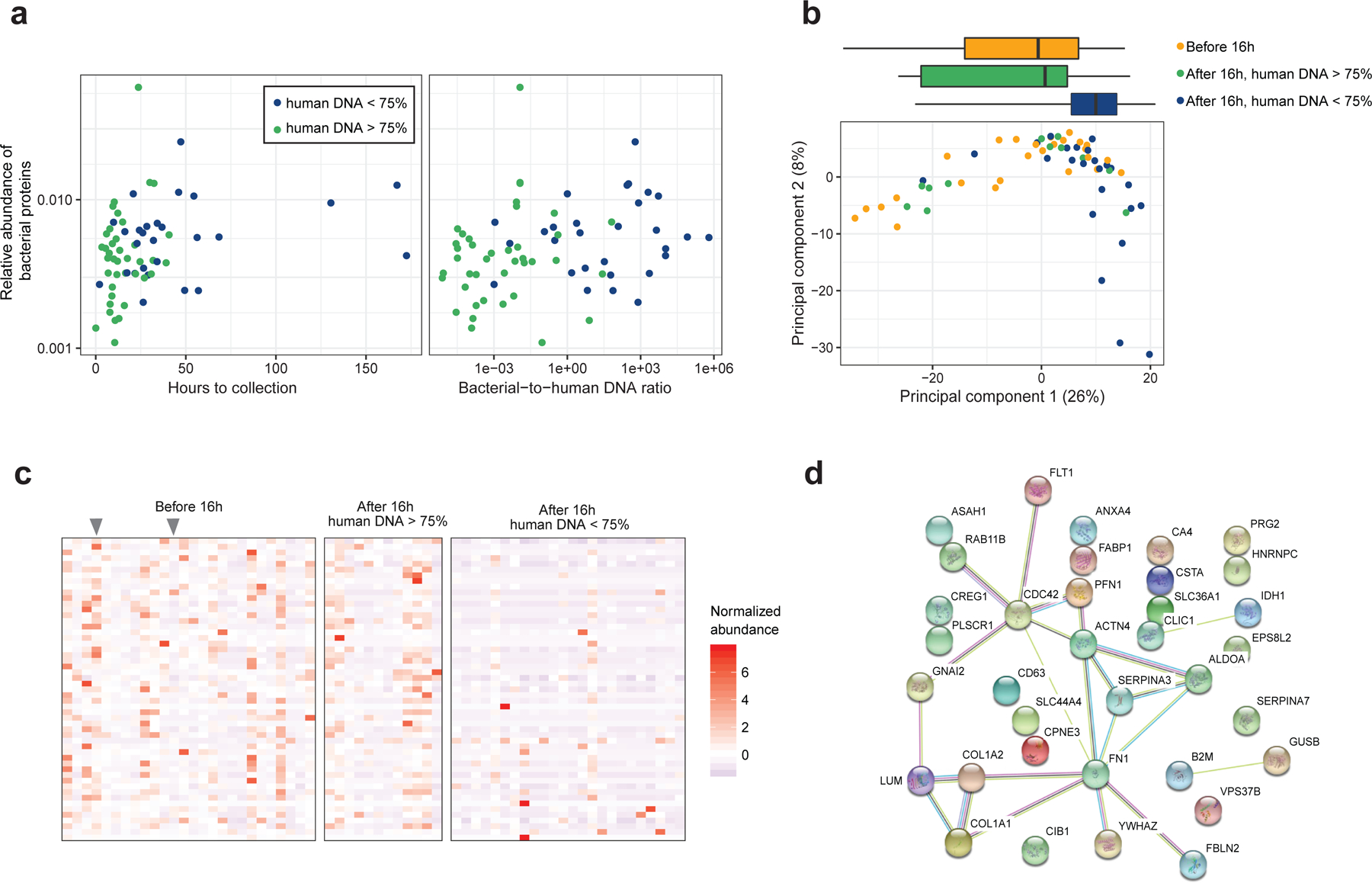
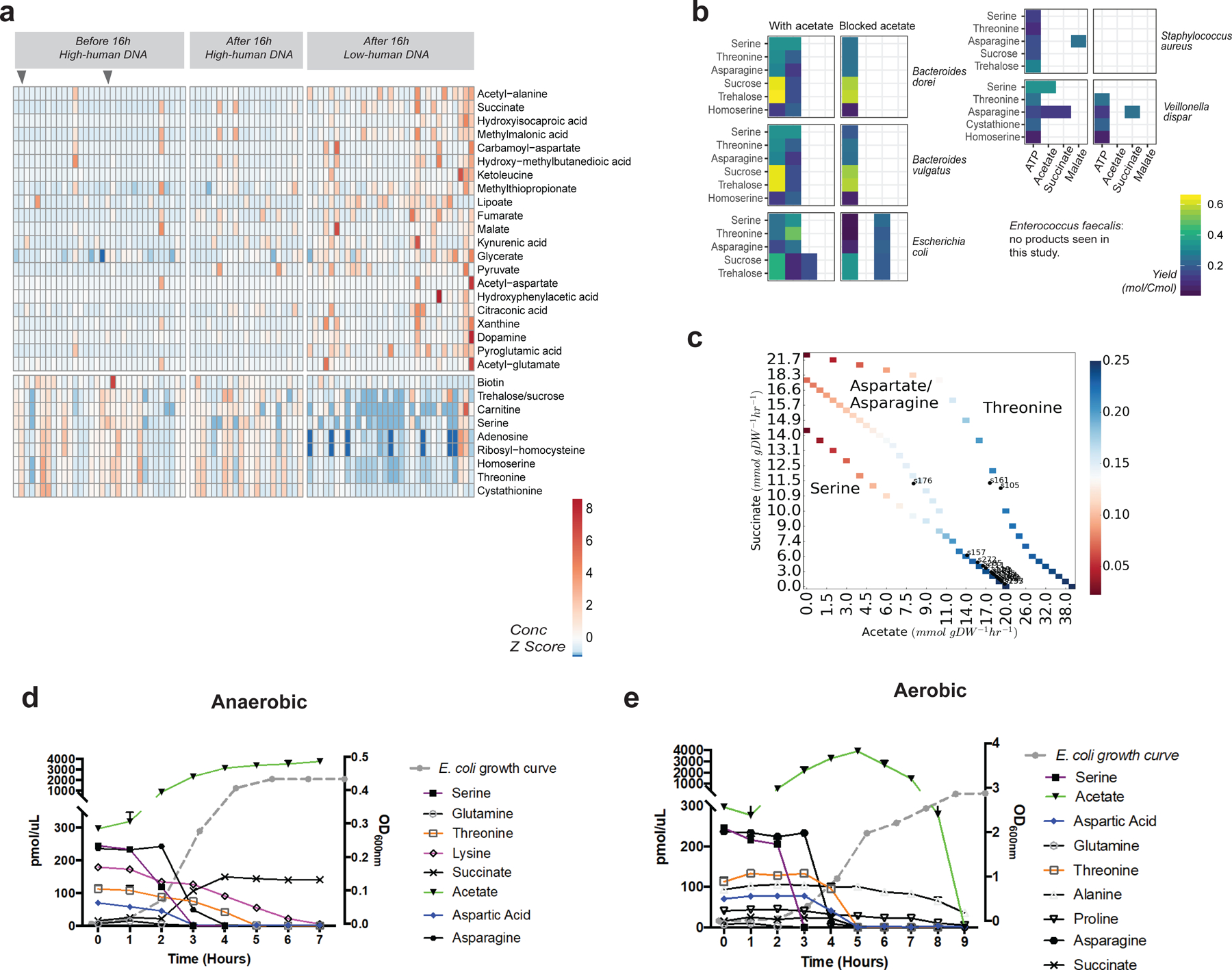
Initial microbial colonization and later succession in the gut of human infants are linked to health and disease later in life. The timing of the appearance of the first gut microbiome, and the consequences for the early life metabolome, are just starting to be defined. Here, we evaluated the gut microbiome, proteome and metabolome in 88 African-American newborns using faecal samples collected in the first few days of life. Gut bacteria became detectable using molecular methods by 16 h after birth. Detailed analysis of the three most common species, Escherichia coli, Enterococcus faecalis and Bacteroides vulgatus, did not suggest a genomic signature for neonatal gut colonization. The appearance of bacteria was associated with reduced abundance of approximately 50 human proteins, decreased levels of free amino acids and an increase in products of bacterial fermentation, including acetate and succinate. Using flux balance modelling and in vitro experiments, we provide evidence that fermentation of amino acids provides a mechanism for the initial growth of E. coli, the most common early colonizer, under anaerobic conditions. These results provide a deep characterization of the first microbes in the human gut and show how the biochemical environment is altered by their appearance.
Conflict of interest statement
Competing Interests
The authors declare no competing interests.
Figures

Comment in
-
Microbial colonization alters neonatal gut metabolome.Nat Microbiol. 2020 Jun;5(6):785-786. doi: 10.1038/s41564-020-0734-9. Nat Microbiol. 2020. PMID: 32467622 No abstract available.
-
What the Editors are reading - Basic science.J Pediatr Urol. 2021 Apr;17(2):271-272. doi: 10.1016/j.jpurol.2020.12.010. Epub 2020 Dec 10. J Pediatr Urol. 2021. PMID: 33358556 No abstract available.
Similar articles
-
The Microbiome and Metabolome of Preterm Infant Stool Are Personalized and Not Driven by Health Outcomes, Including Necrotizing Enterocolitis and Late-Onset Sepsis.mSphere. 2018 Jun 6;3(3):e00104-18. doi: 10.1128/mSphere.00104-18. Print 2018 Jun 27. mSphere. 2018. PMID: 29875143 Free PMC article.
-
Gut Dysbiosis With Bacilli Dominance and Accumulation of Fermentation Products Precedes Late-onset Sepsis in Preterm Infants.Clin Infect Dis. 2019 Jul 2;69(2):268-277. doi: 10.1093/cid/ciy882. Clin Infect Dis. 2019. PMID: 30329017
-
Hospitalized Premature Infants Are Colonized by Related Bacterial Strains with Distinct Proteomic Profiles.mBio. 2018 Apr 10;9(2):e00441-18. doi: 10.1128/mBio.00441-18. mBio. 2018. PMID: 29636439 Free PMC article.
-
Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe.Methods. 2018 Oct 1;149:3-12. doi: 10.1016/j.ymeth.2018.04.029. Epub 2018 Apr 30. Methods. 2018. PMID: 29715508 Review.
-
Early life colonization of the human gut: microbes matter everywhere.Curr Opin Microbiol. 2018 Aug;44:70-78. doi: 10.1016/j.mib.2018.06.003. Epub 2018 Aug 4. Curr Opin Microbiol. 2018. PMID: 30086431 Review.
Cited by
-
Infant Gut Microbial Metagenome Mining of α-l-Fucosidases with Activity on Fucosylated Human Milk Oligosaccharides and Glycoconjugates.Microbiol Spectr. 2022 Aug 31;10(4):e0177522. doi: 10.1128/spectrum.01775-22. Epub 2022 Aug 9. Microbiol Spectr. 2022. PMID: 35943155 Free PMC article.
-
Neonatal Programming of Microbiota Composition: A Plausible Idea That Is Not Supported by the Evidence.Front Microbiol. 2022 Jun 17;13:825942. doi: 10.3389/fmicb.2022.825942. eCollection 2022. Front Microbiol. 2022. PMID: 35783422 Free PMC article. Review.
-
Associations between Dietary Sugar and Fiber with Infant Gut Microbiome Colonization at 6 Mo of Age.J Nutr. 2024 Jan;154(1):152-162. doi: 10.1016/j.tjnut.2023.09.009. Epub 2023 Sep 16. J Nutr. 2024. PMID: 37717629 Free PMC article.
-
Vaginal and neonatal microbiota in pregnant women with preterm premature rupture of membranes and consecutive early onset neonatal sepsis.BMC Med. 2023 Mar 13;21(1):92. doi: 10.1186/s12916-023-02805-x. BMC Med. 2023. PMID: 36907851 Free PMC article.
-
Metabolic profiles of meconium in preeclamptic and normotensive pregnancies.Metabolomics. 2025 Jan 25;21(1):21. doi: 10.1007/s11306-025-02224-4. Metabolomics. 2025. PMID: 39863780 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
