

Sci-fate characterizes the dynamics of gene expression in single cells
- PMID: 32284584
- PMCID: PMC7416490
- DOI: 10.1038/s41587-020-0480-9
Sci-fate characterizes the dynamics of gene expression in single cells
Abstract
Gene expression programs change over time, differentiation and development, and in response to stimuli. However, nearly all techniques for profiling gene expression in single cells do not directly capture transcriptional dynamics. In the present study, we present a method for combined single-cell combinatorial indexing and messenger RNA labeling (sci-fate), which uses combinatorial cell indexing and 4-thiouridine labeling of newly synthesized mRNA to concurrently profile the whole and newly synthesized transcriptome in each of many single cells. We used sci-fate to study the cortisol response in >6,000 single cultured cells. From these data, we quantified the dynamics of the cell cycle and glucocorticoid receptor activation, and explored their intersection. Finally, we developed software to infer and analyze cell-state transitions. We anticipate that sci-fate will be broadly applicable to quantitatively characterize transcriptional dynamics in diverse systems.
Figures
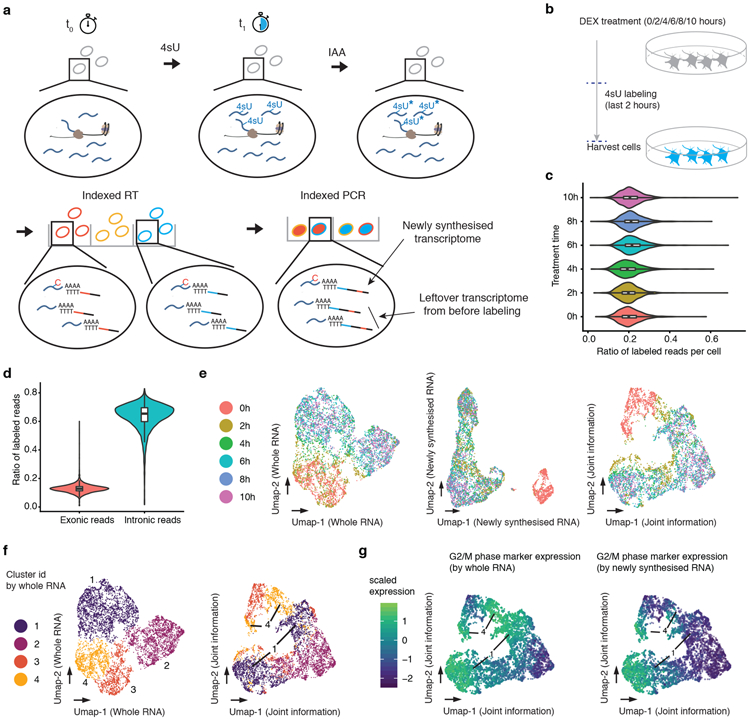
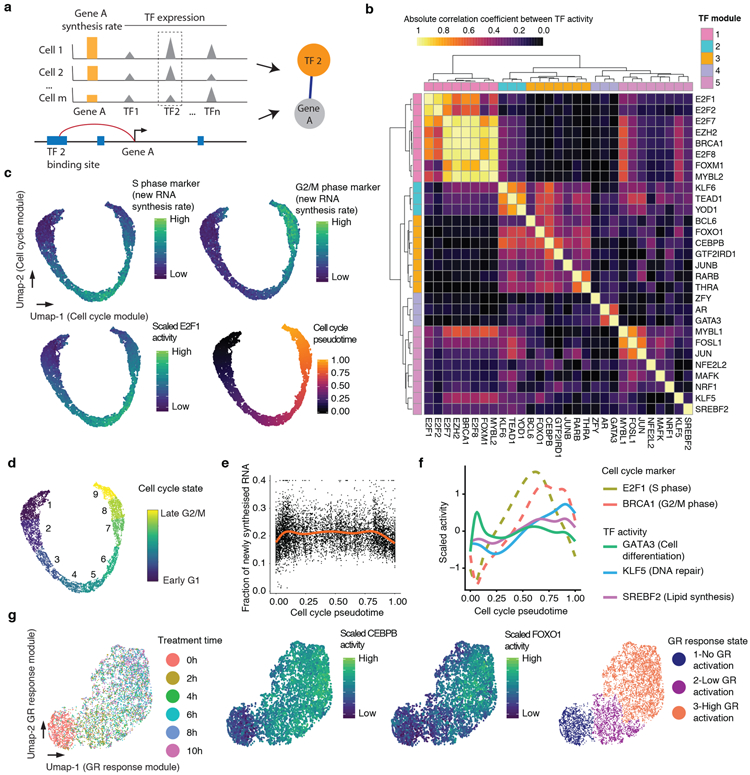
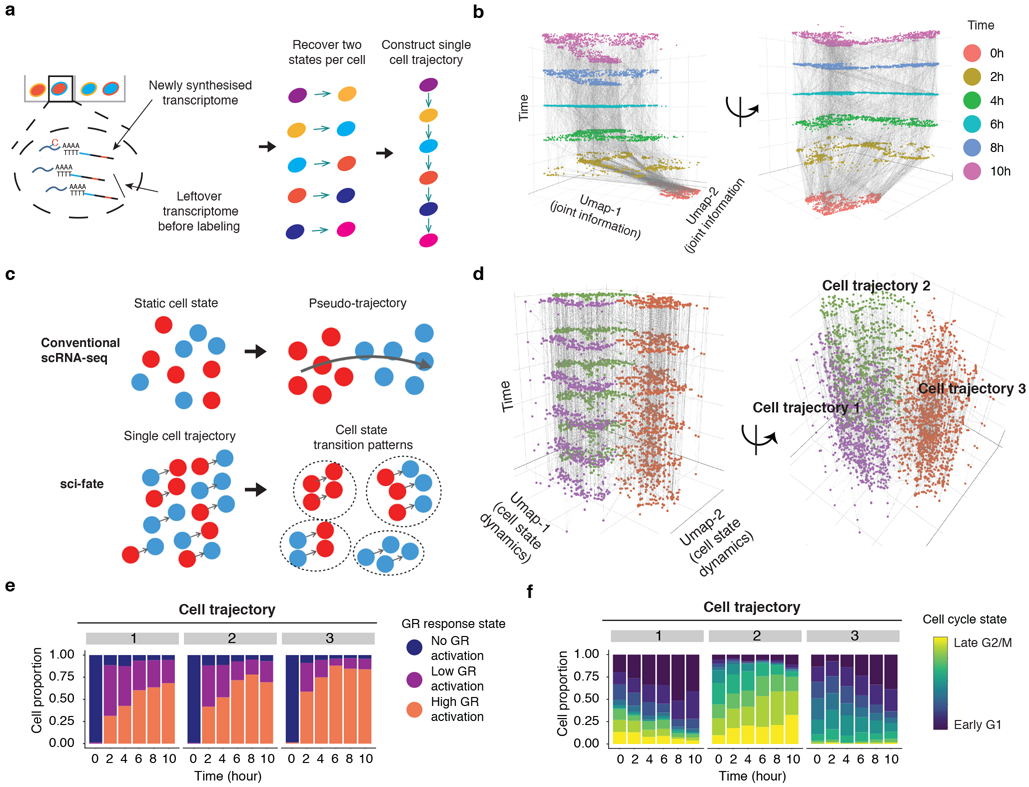
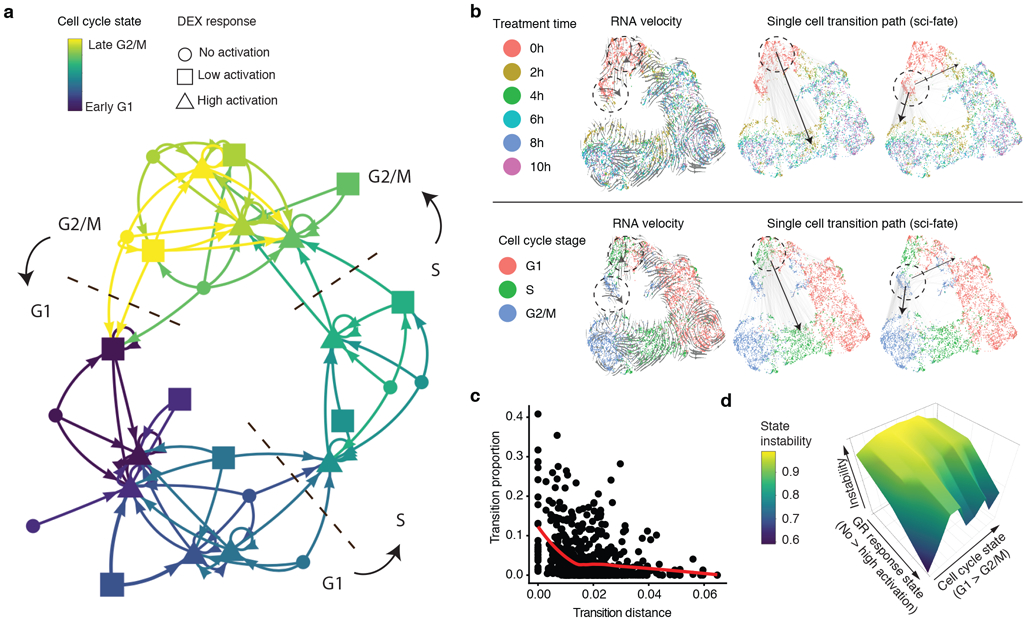
References
-
- Haghverdi L, Büttner M, Wolf FA, Buettner F & Theis FJ Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848 (2016). - PubMed
Methods-only References
-
- Lindenbaum P JVarkit: java-based utilities for Bioinformatics. figshare (2015).
-
- FelixKrueger. FelixKrueger/TrimGalore. GitHub https://github.com/FelixKrueger/TrimGalore.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
