

Idiopathic pulmonary arterial hypertension and co-existing lung disease: is this a new phenotype?
- PMID: 32284847
- PMCID: PMC7132795
- DOI: 10.1177/2045894020914851
Idiopathic pulmonary arterial hypertension and co-existing lung disease: is this a new phenotype?
Abstract
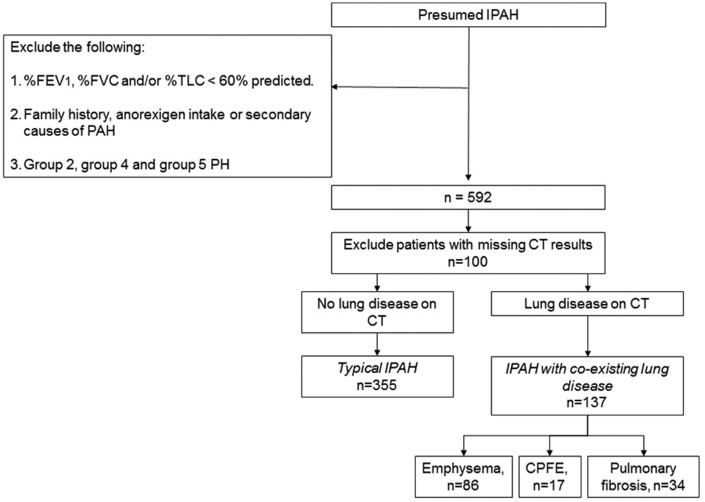
Patients classified as idiopathic pulmonary arterial hypertension (defined as Group 1 on European Respiratory Society (ERS)/European Cardiac Society (ESC) criteria) may have evidence of minor co-existing lung disease on thoracic computed tomography. We hypothesised that these idiopathic pulmonary arterial hypertension patients (IPAH lung disease ) are a separate subgroup of idiopathic pulmonary arterial hypertension with different phenotype and outcome compared with idiopathic pulmonary arterial hypertension patients without co-existing lung disease (IPAH no lung disease ). Patients with 'IPAH lung disease ' have been eligible for all clinical trials of Group 1 patients because they have normal clinical examination and normal spirometry but we wondered whether they responded to treatment and had similar survival to patients with 'IPAH no lung disease '. We described the outcome of the cohort of patients with 'IPAH no lung disease ' in a previous paper. Here, we have compared incident 'IPAH lung disease ' patients with 'IPAH no lung disease ' patients diagnosed concurrently in all eight Pulmonary Hypertension centres in the UK and Ireland between 2001-2009. Compared with 'IPAH no lung disease ' (n = 355), 'IPAH lung disease ' patients (n = 137) were older, less obese, predominantly male, more likely to be current/ex-smokers and had lower six-minute walk distance, lower % predicted diffusion capacity for carbon monoxide, lower mean pulmonary arterial pressure and lower pulmonary vascular resistance index. After three months of pulmonary hypertension-targeted treatment, six-minute walk distance improved equally in 'IPAH lung disease ' and 'IPAH no lung disease '. However, survival of 'IPAH lung disease ' was lower than 'IPAH no lung disease ' (one year survival: 72% compared with 93%). This survival was significantly worse in 'IPAH lung disease ' even after adjusting for age, gender, smoking history, comorbidities and haemodynamics. 'IPAH lung disease ' patients had similar short-term improvement in six-minute walk distance with anti-pulmonary arterial hypertension therapy but worse survival compared with 'IPAH no lung disease ' patients. This suggests that 'IPAH lung disease ' are a separate phenotype and should not be lumped with 'IPAH no lung disease ' in clinical trials of Group 1 pulmonary arterial hypertension.
Keywords: idiopathic pulmonary arterial hypertension (IPAH); lung disease; survival; treatment response.
© The Author(s) 2020.
Figures
References
-
- Ling Y, Johnson MK, Kiely DG, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension. Results from the Pulmonary Hypertension Registry of the United Kingdom and Ireland. Am J Resp Crit Care Med 2012; 186: 790–796. - PubMed
-
- Hoeper MM, Huscher D, Ghofrani HA, et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: results from the COMPERA registry. Int J Cardiol 2013; 168: 871–880. - PubMed
-
- Trip P, Nossent EJ, de Man FS, et al. Severely reduced diffusion capacity in idiopathic pulmonary arterial hypertension: patient characteristics and treatment responses. Eur Respir J 2013; 42: 1575–1585. - PubMed
-
- Clark KD, Wardrobe-Wong N, Elliott JJ, et al. Are emphysema and airflow obstruction found together? Chest 2001; 120: 743–747. - PubMed
LinkOut - more resources
Full Text Sources
