

The hypertrophic cardiomyopathy mutations R403Q and R663H increase the number of myosin heads available to interact with actin
- PMID: 32284968
- PMCID: PMC7124958
- DOI: 10.1126/sciadv.aax0069
The hypertrophic cardiomyopathy mutations R403Q and R663H increase the number of myosin heads available to interact with actin
Abstract
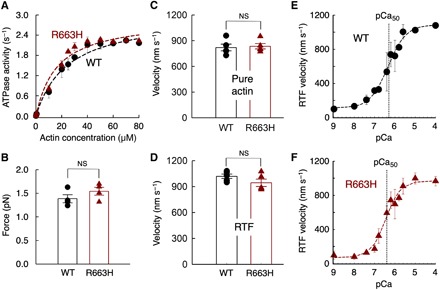
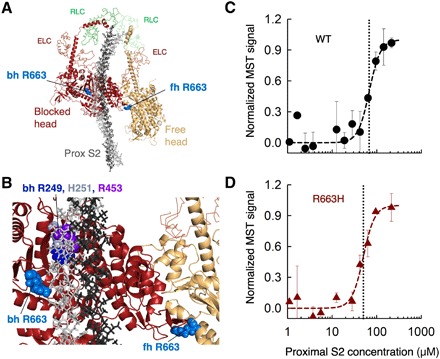
Hypertrophic cardiomyopathy (HCM) mutations in β-cardiac myosin and myosin binding protein-C (MyBP-C) lead to hypercontractility of the heart, an early hallmark of HCM. We show that hypercontractility caused by the HCM-causing mutation R663H cannot be explained by changes in fundamental myosin contractile parameters, much like the HCM-causing mutation R403Q. Using enzymatic assays with purified human β-cardiac myosin, we provide evidence that both mutations cause hypercontractility by increasing the number of functionally accessible myosin heads. We also demonstrate that the myosin mutation R403Q, but not R663H, ablates the binding of myosin with the C0-C7 fragment of MyBP-C. Furthermore, addition of C0-C7 decreases the wild-type myosin basal ATPase single turnover rate, while the mutants do not show a similar reduction. These data suggest that a primary mechanism of action for these mutations is to increase the number of myosin heads functionally available for interaction with actin, which could contribute to hypercontractility.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures


References
-
- Semsarian C., Ingles J., Maron M. S., Maron B. J., New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
