

Minimum epistasis interpolation for sequence-function relationships
- PMID: 32286265
- PMCID: PMC7156698
- DOI: 10.1038/s41467-020-15512-5
Minimum epistasis interpolation for sequence-function relationships
Abstract
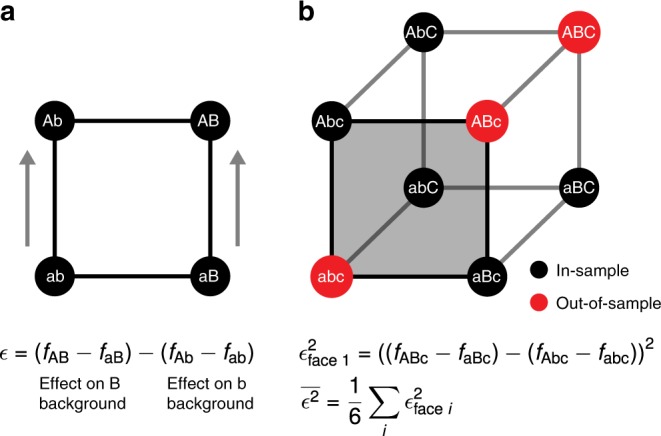
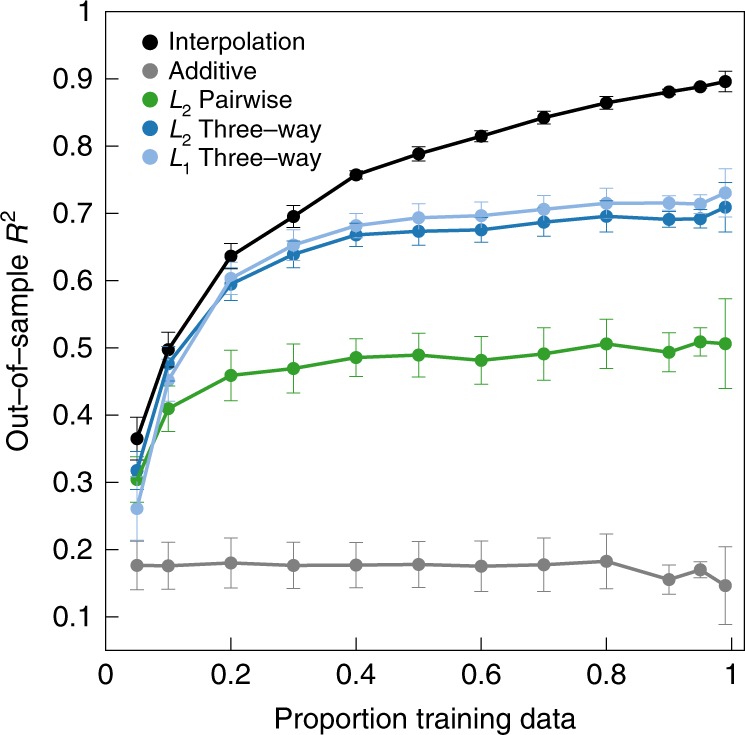
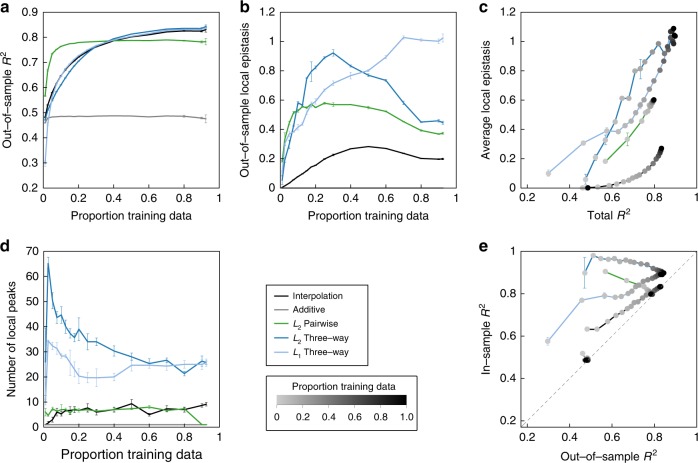
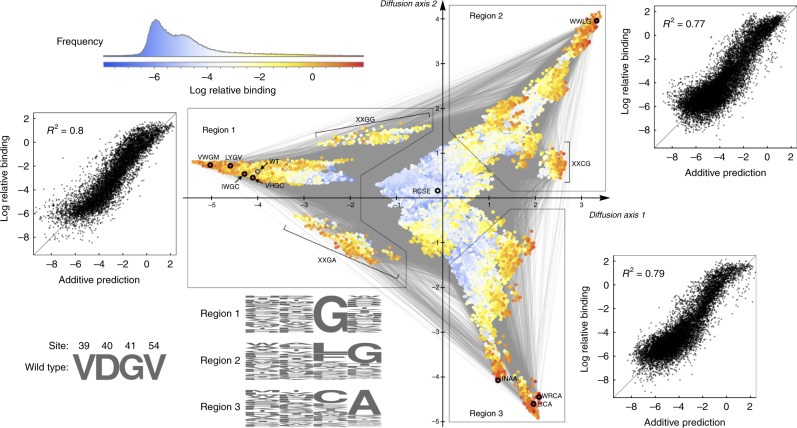
Massively parallel phenotyping assays have provided unprecedented insight into how multiple mutations combine to determine biological function. While such assays can measure phenotypes for thousands to millions of genotypes in a single experiment, in practice these measurements are not exhaustive, so that there is a need for techniques to impute values for genotypes whose phenotypes have not been directly assayed. Here, we present an imputation method based on inferring the least epistatic possible sequence-function relationship compatible with the data. In particular, we infer the reconstruction where mutational effects change as little as possible across adjacent genetic backgrounds. The resulting models can capture complex higher-order genetic interactions near the data, but approach additivity where data is sparse or absent. We apply the method to high-throughput transcription factor binding assays and use it to explore a fitness landscape for protein G.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
