

Thermodynamic and Evolutionary Coupling between the Native and Amyloid State of Globular Proteins
- PMID: 32294448
- PMCID: PMC7175379
- DOI: 10.1016/j.celrep.2020.03.076
Thermodynamic and Evolutionary Coupling between the Native and Amyloid State of Globular Proteins
Abstract
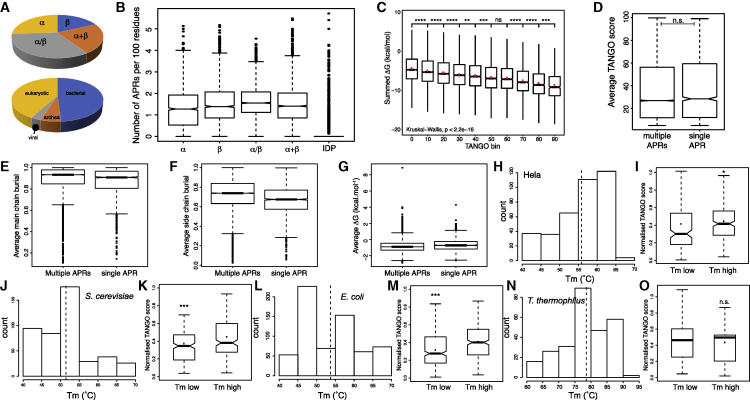
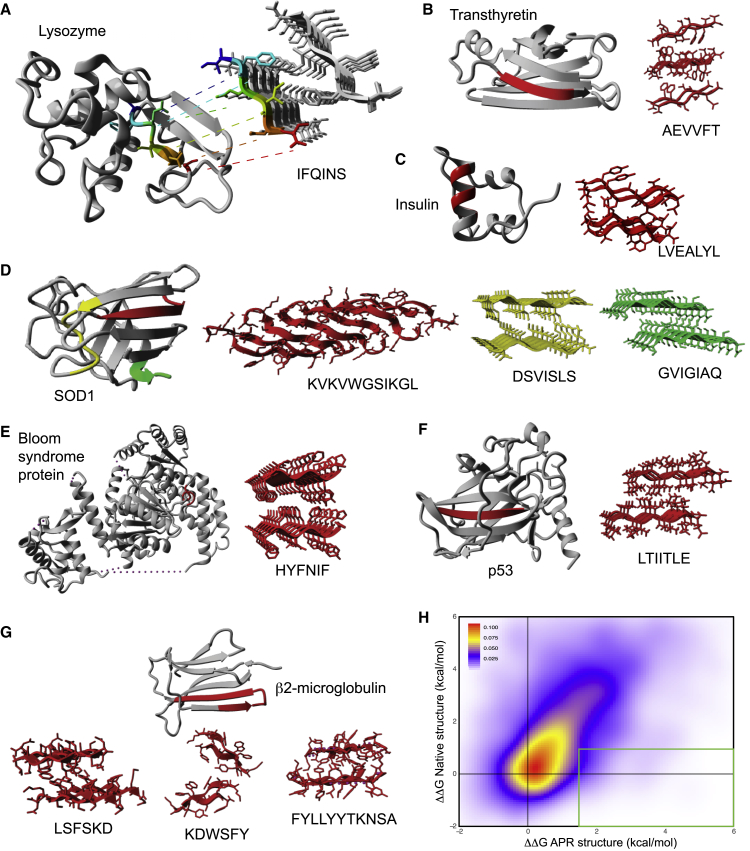
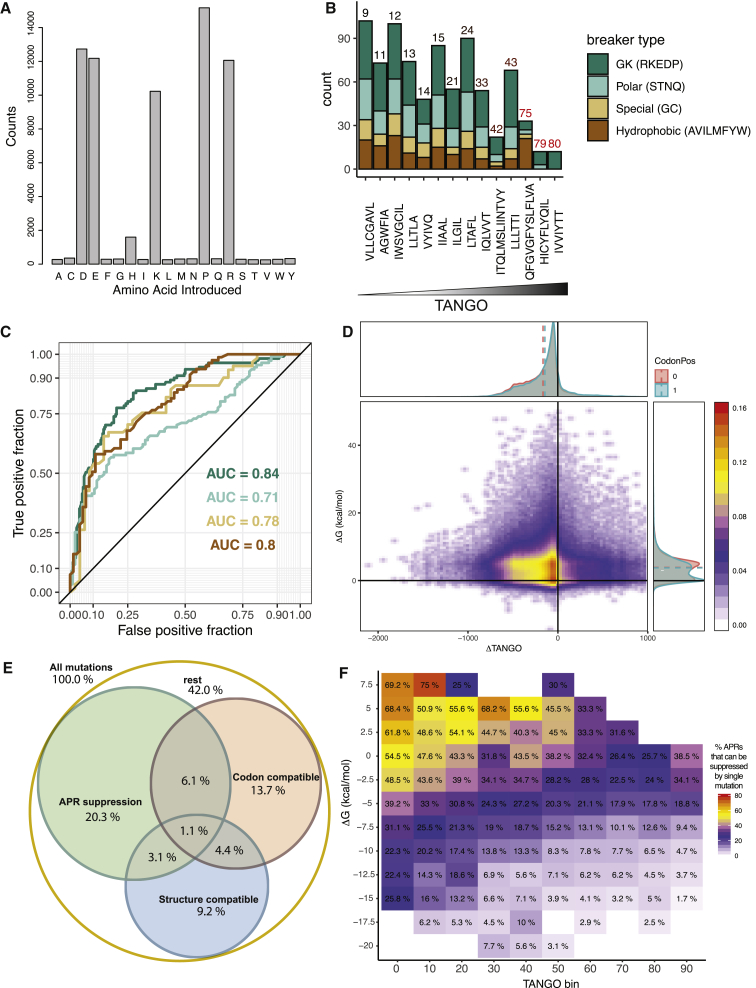
The amyloid-like aggregation propensity present in most globular proteins is generally considered to be a secondary side effect resulting from the requirements of protein stability. Here, we demonstrate, however, that mutations in the globular and amyloid state are thermodynamically correlated rather than simply associated. In addition, we show that the standard genetic code couples this structural correlation into a tight evolutionary relationship. We illustrate the extent of this evolutionary entanglement of amyloid propensity and globular protein stability. Suppressing a 600-Ma-conserved amyloidogenic segment in the p53 core domain fold is structurally feasible but requires 7-bp substitutions to concomitantly introduce two aggregation-suppressing and three stabilizing amino acid mutations. We speculate that, rather than being a corollary of protein evolution, it is equally plausible that positive selection for amyloid structure could have been a driver for the emergence of globular protein structure.
Keywords: amyloid; evolution; protein folding; protein stability.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests F.R. and J.S. are scientific founders of Aelin Therapeutics and members of its scientific advisory board.
Figures
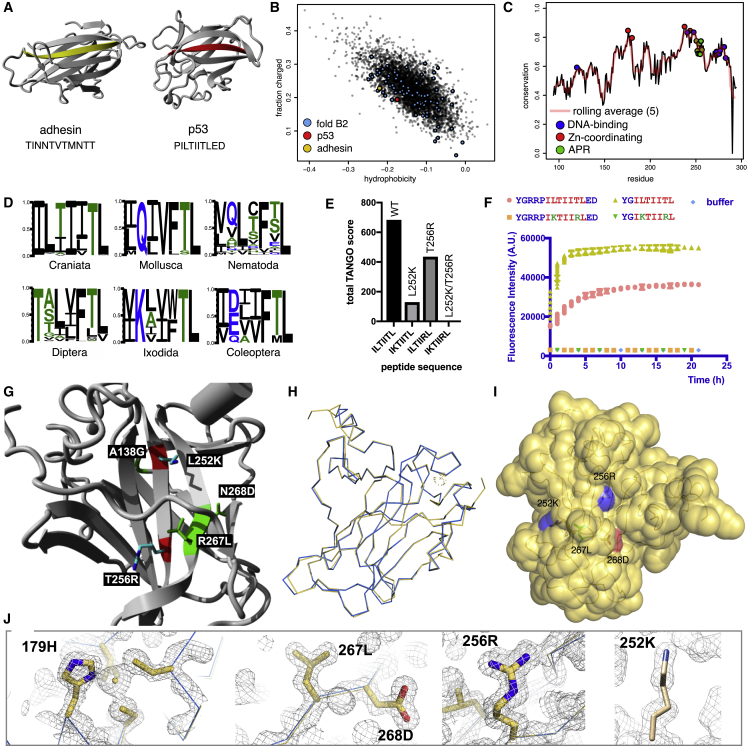
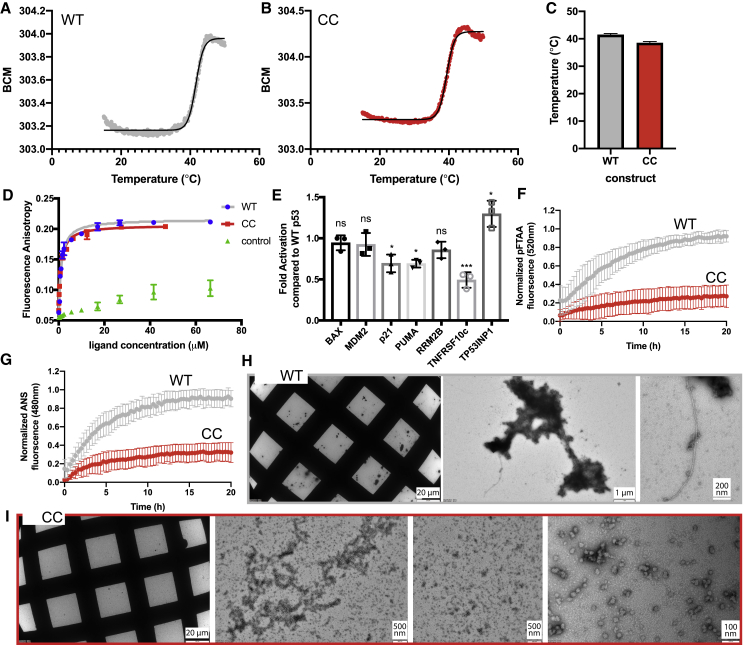
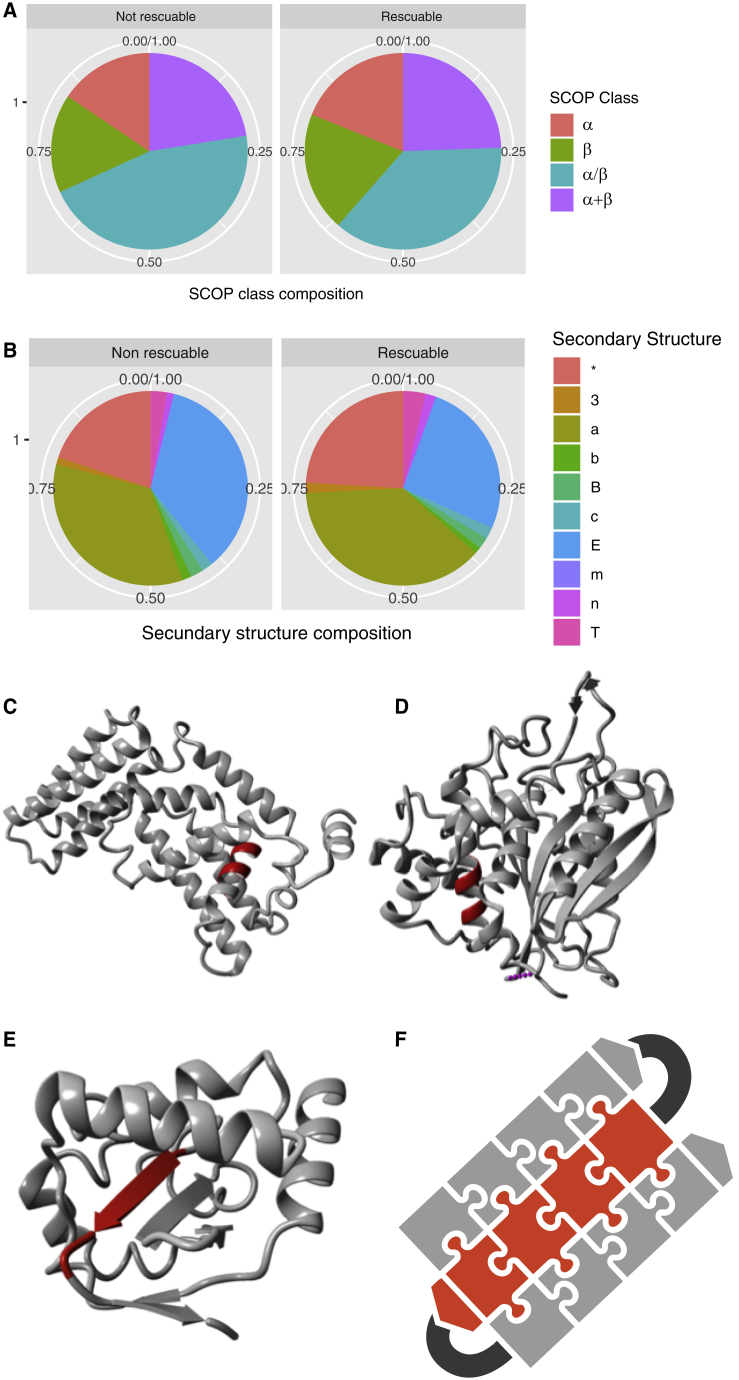
References
-
- Al-Garawi Z.S., McIntosh B.A., Neill-Hall D., Hatimy A.A., Sweet S.M., Bagley M.C., Serpell L.C. The amyloid architecture provides a scaffold for enzyme-like catalysts. Nanoscale. 2017;9:10773–10783. - PubMed
-
- Anfinsen C.B. Principles that govern the folding of protein chains. Science. 1973;181:223–230. - PubMed
-
- Anfinsen C.B., Haber E. Studies on the reduction and re-formation of protein disulfide bonds. J. Biol. Chem. 1961;236:1361–1363. - PubMed
-
- Bada J.L. New insights into prebiotic chemistry from Stanley Miller’s spark discharge experiments. Chem. Soc. Rev. 2013;42:2186–2196. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
