

American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults
- PMID: 32298430
- PMCID: PMC7189278
- DOI: 10.1182/bloodadvances.2019001142
American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults
Abstract
Background: Central nervous system (CNS) complications are among the most common, devastating sequelae of sickle cell disease (SCD) occurring throughout the lifespan.
Objective: These evidence-based guidelines of the American Society of Hematology are intended to support the SCD community in decisions about prevention, diagnosis, and treatment of the most common neurological morbidities in SCD.
Methods: The Mayo Evidence-Based Practice Research Program supported the guideline development process, including updating or performing systematic evidence reviews. The panel used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, including GRADE evidence-to-decision frameworks, to assess evidence and make recommendations.
Results: The panel placed a higher value on maintaining cognitive function than on being alive with significantly less than baseline cognitive function. The panel developed 19 recommendations with evidence-based strategies to prevent, diagnose, and treat CNS complications of SCD in low-middle- and high-income settings.
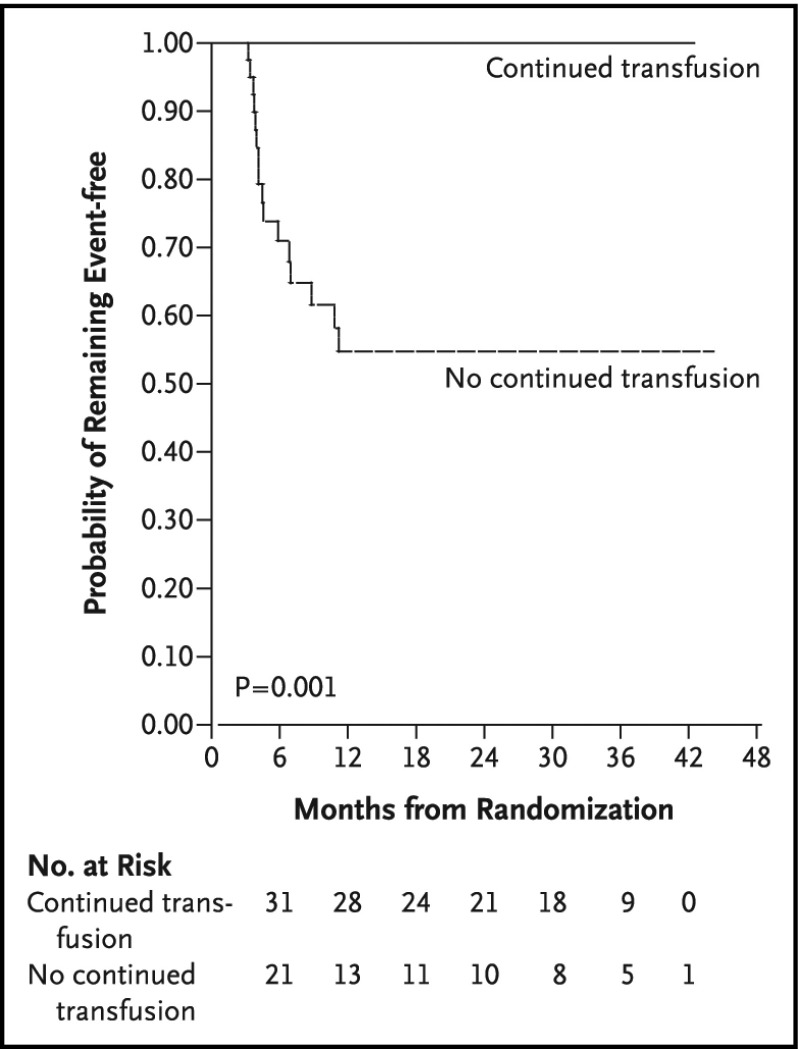
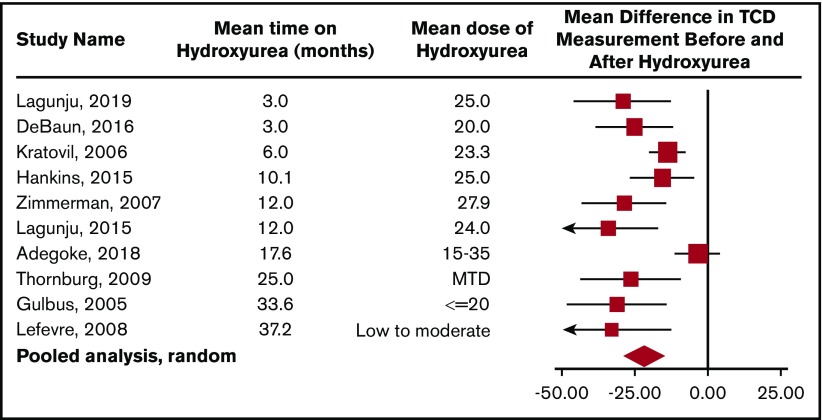
Conclusions: Three of 19 recommendations immediately impact clinical care. These recommendations include: use of transcranial Doppler ultrasound screening and hydroxyurea for primary stroke prevention in children with hemoglobin SS (HbSS) and hemoglobin Sβ0 (HbSβ0) thalassemia living in low-middle-income settings; surveillance for developmental delay, cognitive impairments, and neurodevelopmental disorders in children; and use of magnetic resonance imaging of the brain without sedation to detect silent cerebral infarcts at least once in early-school-age children and once in adults with HbSS or HbSβ0 thalassemia. Individuals with SCD, their family members, and clinicians should become aware of and implement these recommendations to reduce the burden of CNS complications in children and adults with SCD.
Keywords: Anemia-Clinical: Sickle cell anemia; RED CELLS.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: All authors were members of the guideline panel, the systematic review team, or both. As such, they completed a disclosure-of-interest form, which was reviewed by ASH and is available as supplemental Files 2 and 3.
Figures
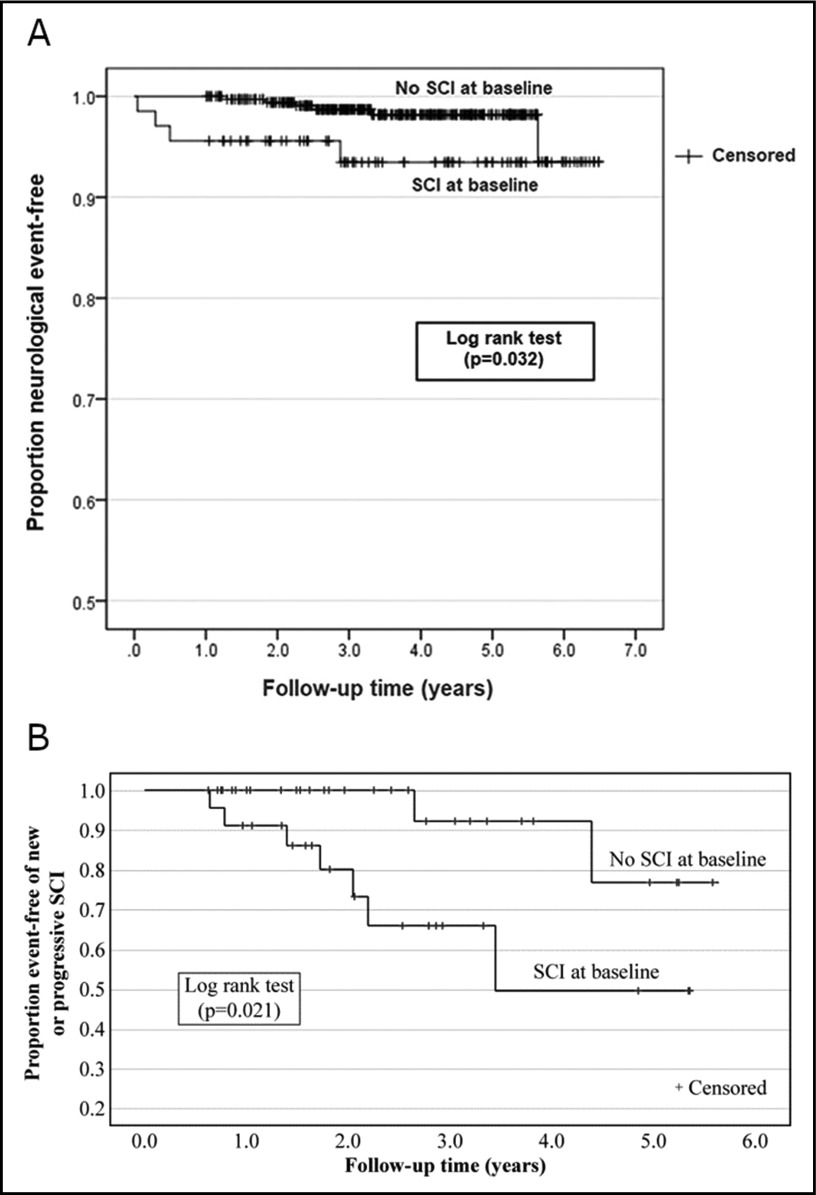
Comment in
-
Neurocognitive screening in sickle cell disease.Pediatr Blood Cancer. 2022 Sep;69(9):e29861. doi: 10.1002/pbc.29861. Epub 2022 Jul 5. Pediatr Blood Cancer. 2022. PMID: 35716349 No abstract available.
References
-
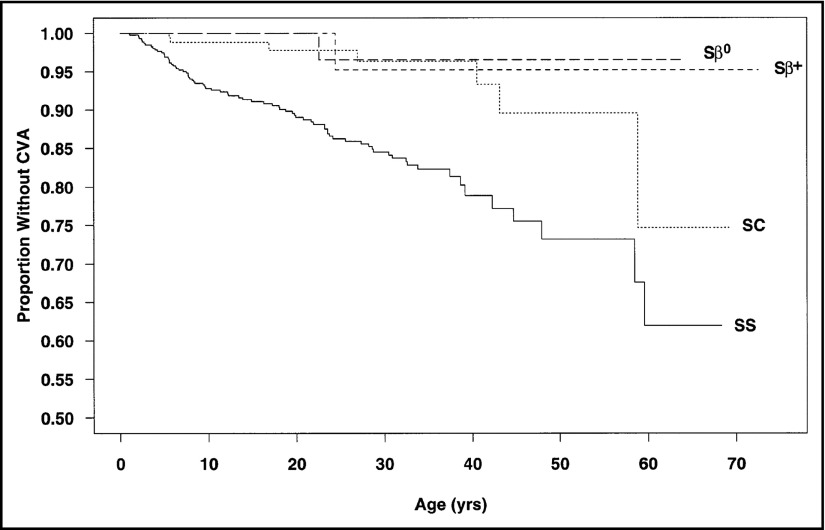
- Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. . Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288-294. - PubMed
-
- Bernaudin F, Verlhac S, Arnaud C, et al. . Chronic and acute anemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood. 2015;125(10):1653-1661. - PubMed
-
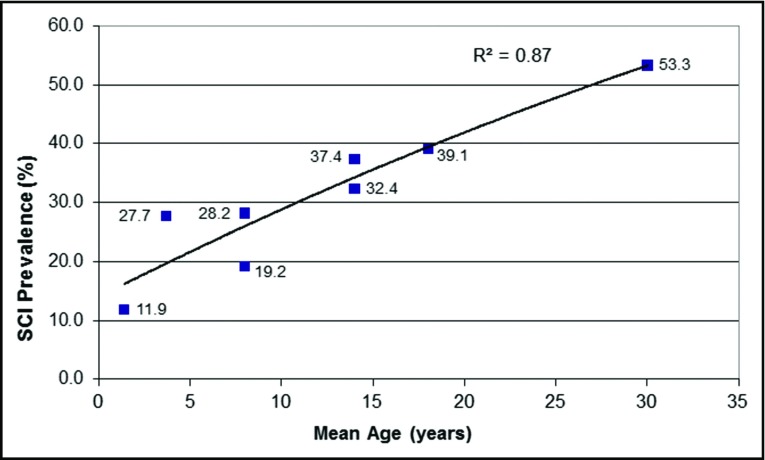
- Kassim AA, Pruthi S, Day M, et al. . Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood. 2016;127(16):2038-2040. - PubMed
-
- Glauser TA, Siegel MJ, Lee BC, DeBaun MR. Accuracy of neurologic examination and history in detecting evidence of MRI-diagnosed cerebral infarctions in children with sickle cell hemoglobinopathy. J Child Neurol. 1995;10(2):88-92. - PubMed
