

High-resolution probing of early events in amyloid-β aggregation related to Alzheimer's disease
- PMID: 32300761
- PMCID: PMC7254607
- DOI: 10.1039/d0cc01551b
High-resolution probing of early events in amyloid-β aggregation related to Alzheimer's disease
Abstract
In Alzheimer's disease (AD), soluble oligomers of amyloid-β (Aβ) are emerging as a crucial entity in driving disease progression as compared to insoluble amyloid deposits. The lacuna in establishing the structure to function relationship for Aβ oligomers prevents the development of an effective treatment for AD. While the transient and heterogeneous properties of Aβ oligomers impose many challenges for structural investigation, an effective use of a combination of NMR techniques has successfully identified and characterized them at atomic-resolution. Here, we review the successful utilization of solution and solid-state NMR techniques to probe the aggregation and structures of small and large oligomers of Aβ. Biophysical studies utilizing the commonly used solution and 19F based NMR experiments to identify the formation of small size early intermediates and to obtain their structures, and dock-lock mechanism of fiber growth at atomic-resolution are discussed. In addition, the use of proton-detected magic angle spinning (MAS) solid-state NMR experiments to obtain high-resolution insights into the aggregation pathways and structures of large oligomers and other aggregates is also presented. We expect these NMR based studies to be valuable for real-time monitoring of the depletion of monomers and the formation of toxic oligomers and high-order aggregates under a variety of conditions, and to solve the high-resolution structures of small and large size oligomers for most amyloid proteins, and therefore to develop inhibitors and drugs.
Conflict of interest statement
Conflicts of interest
There are no conflicts to declare.
Figures
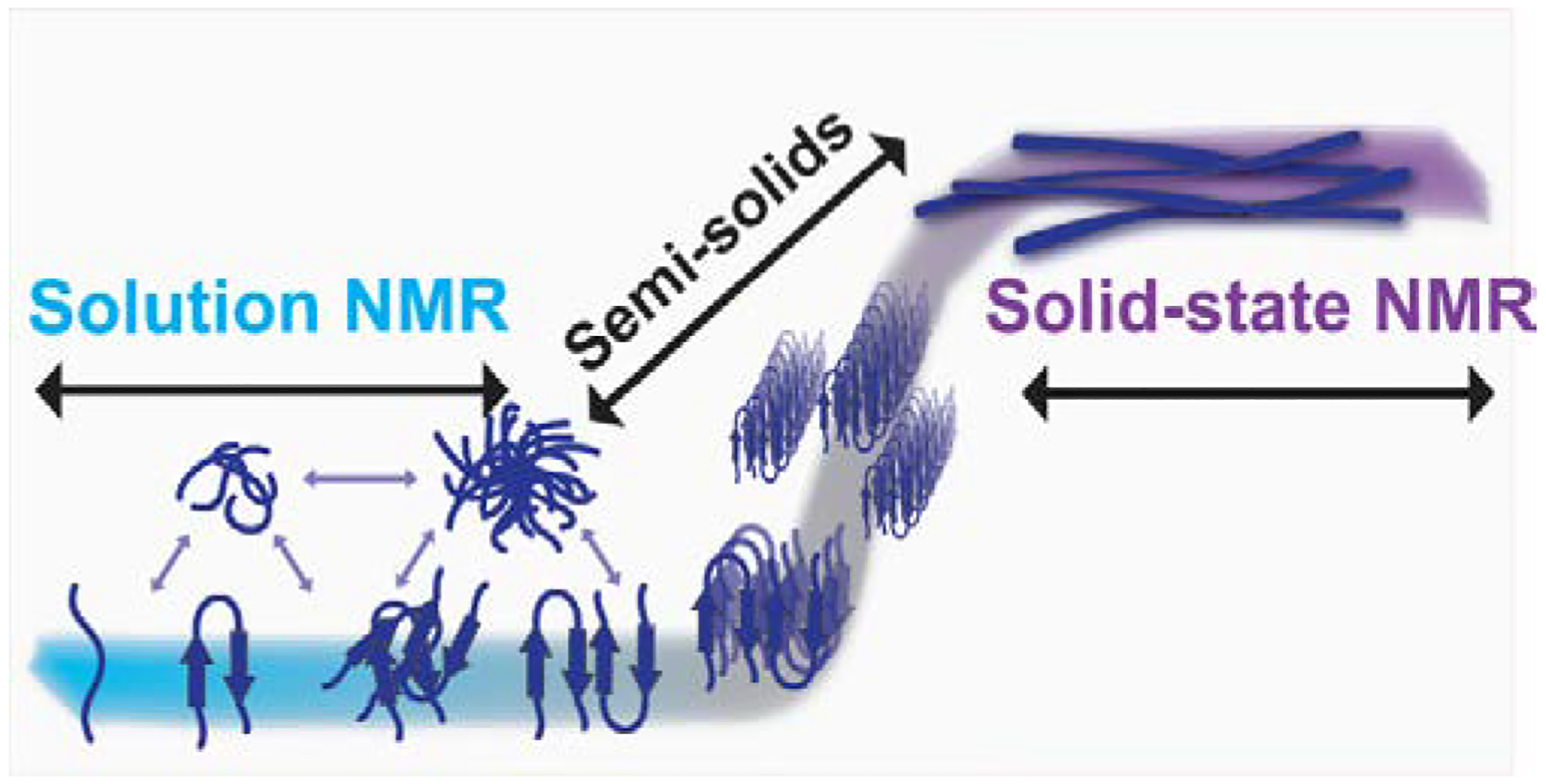
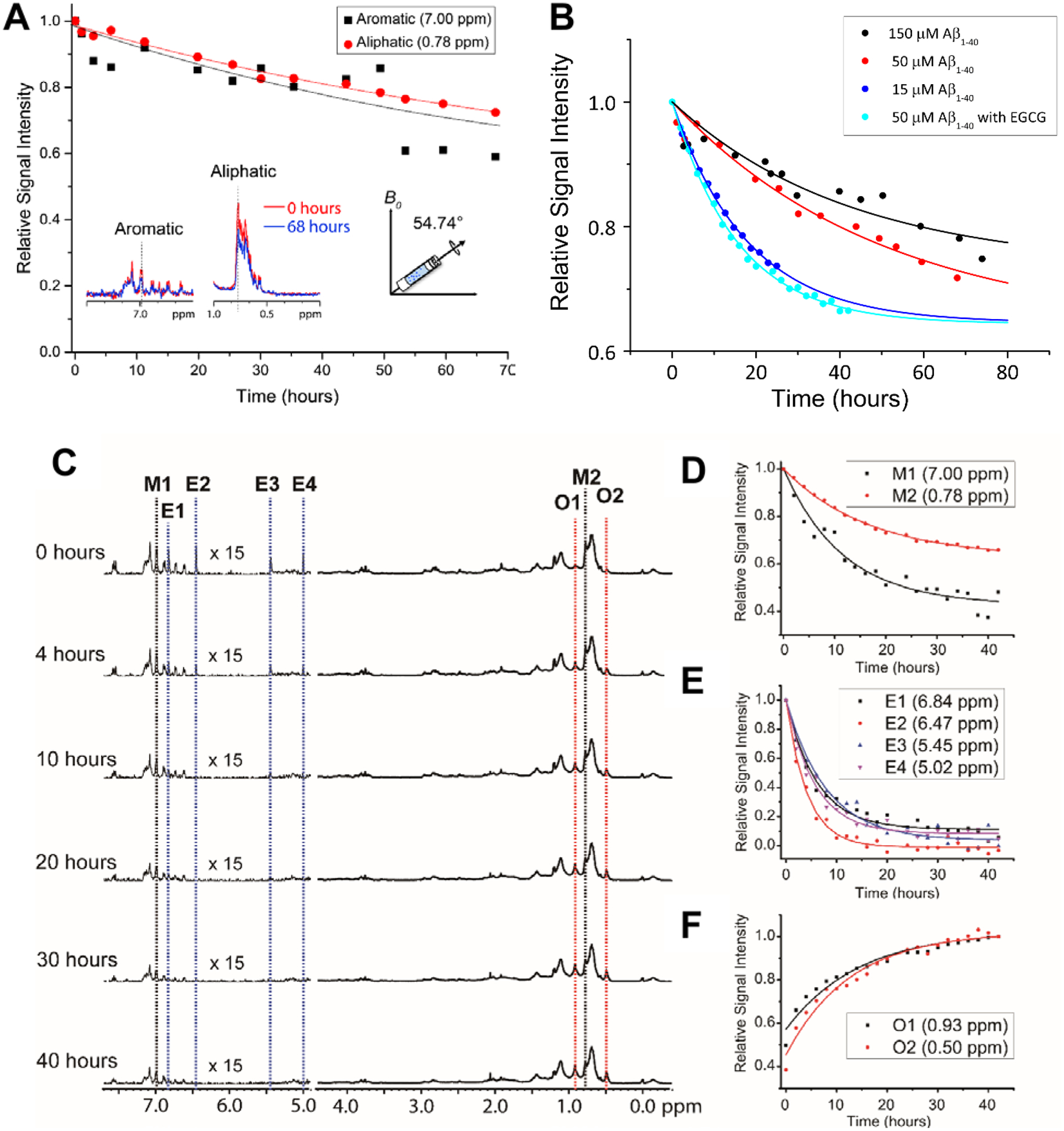
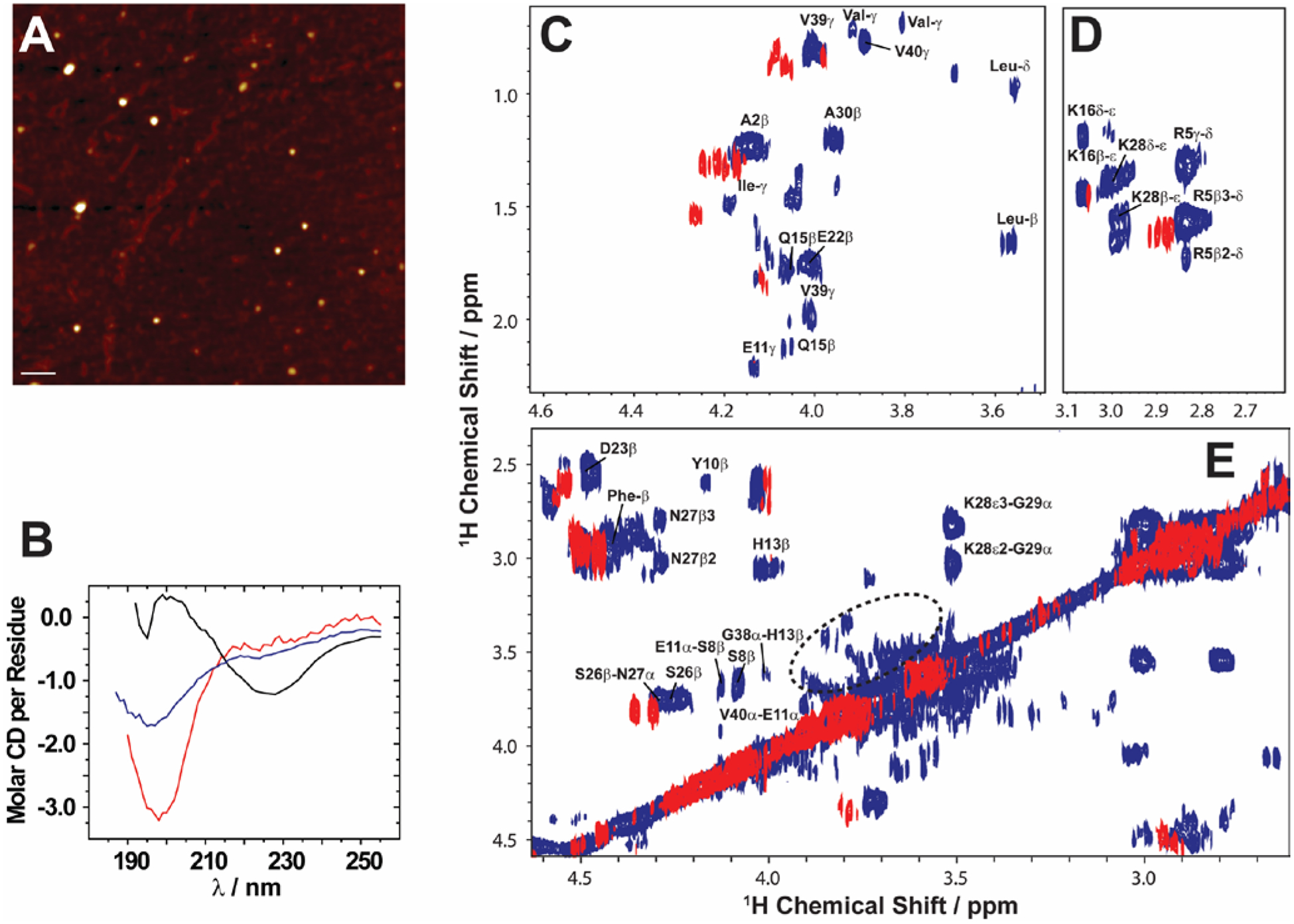
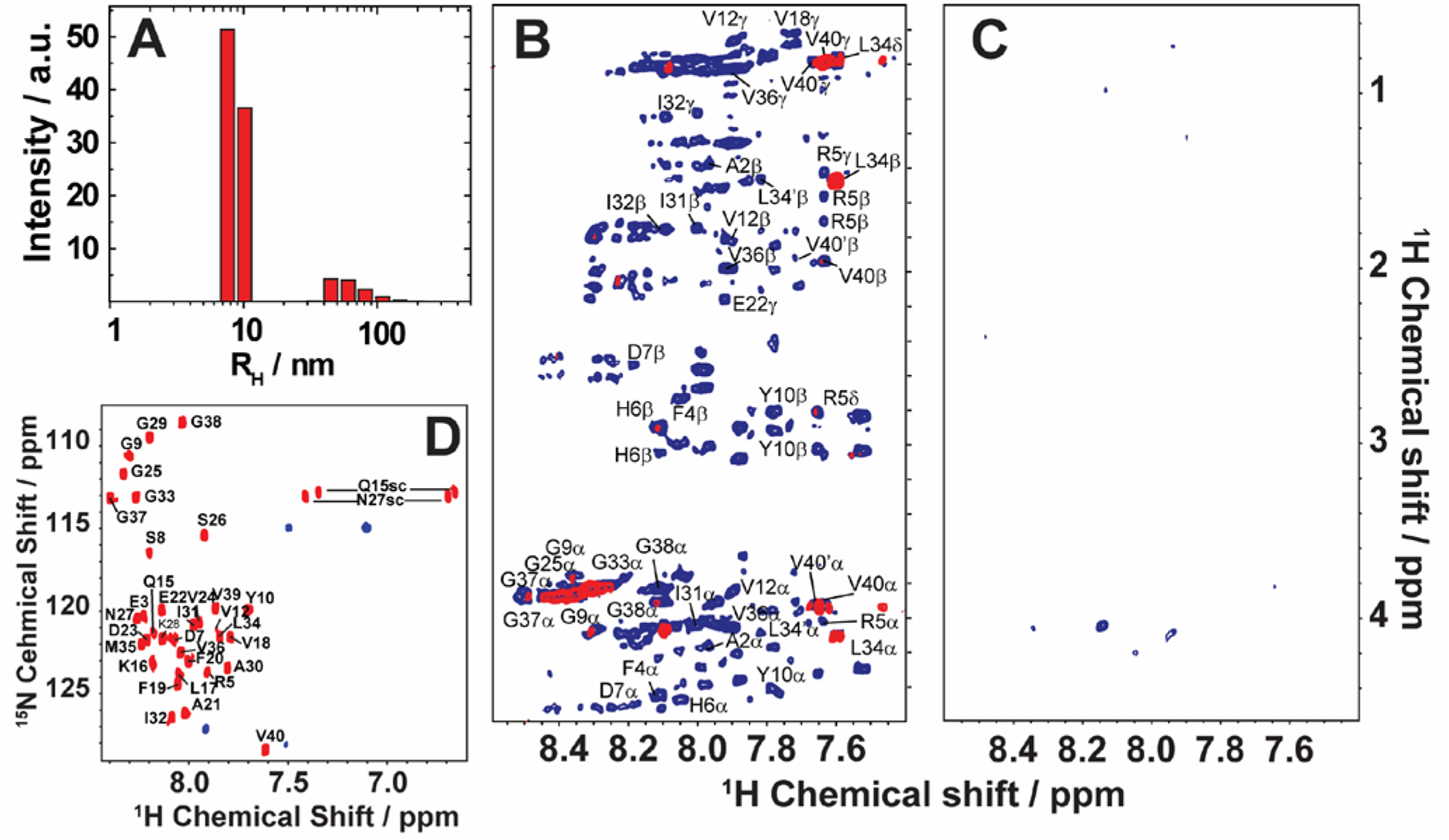
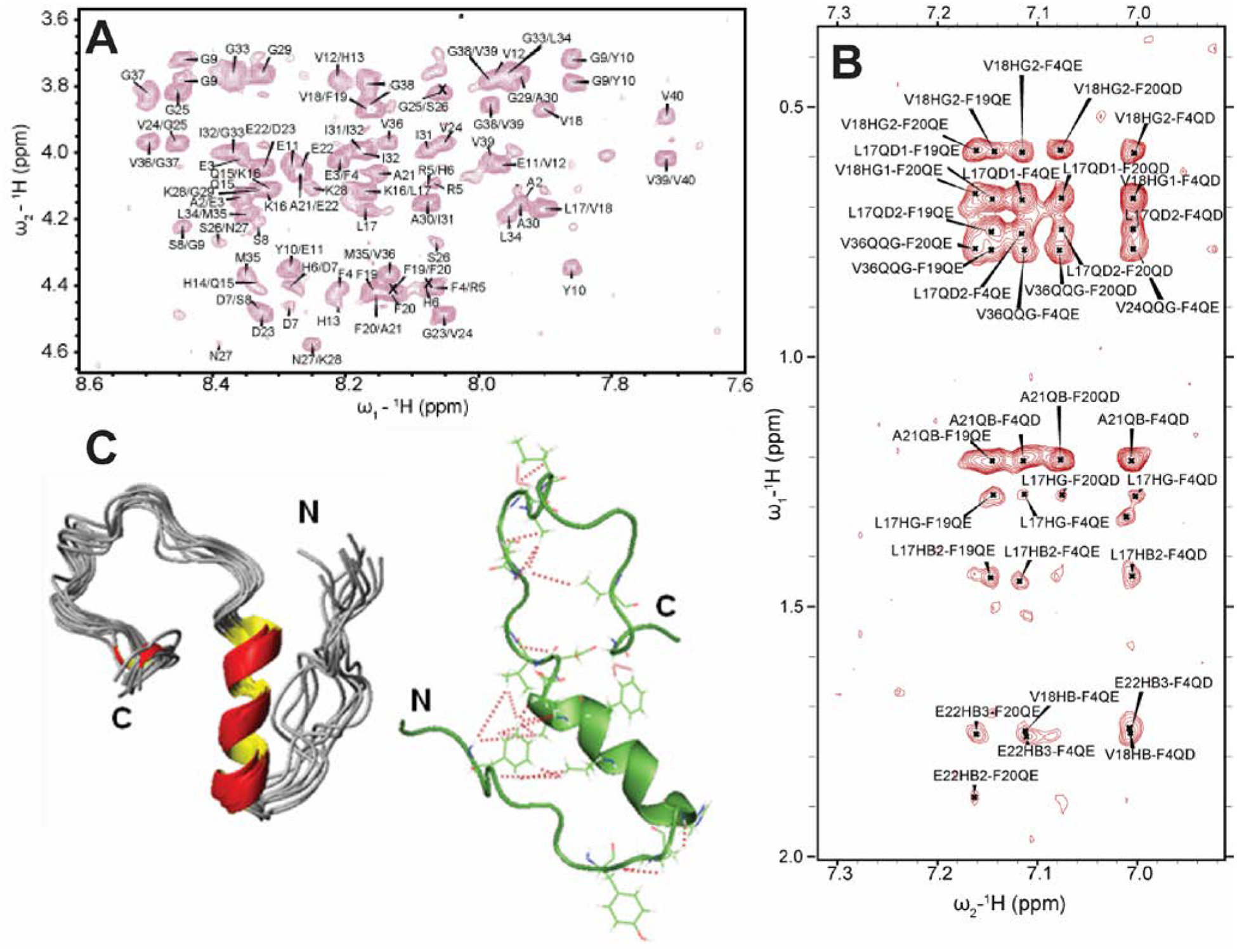
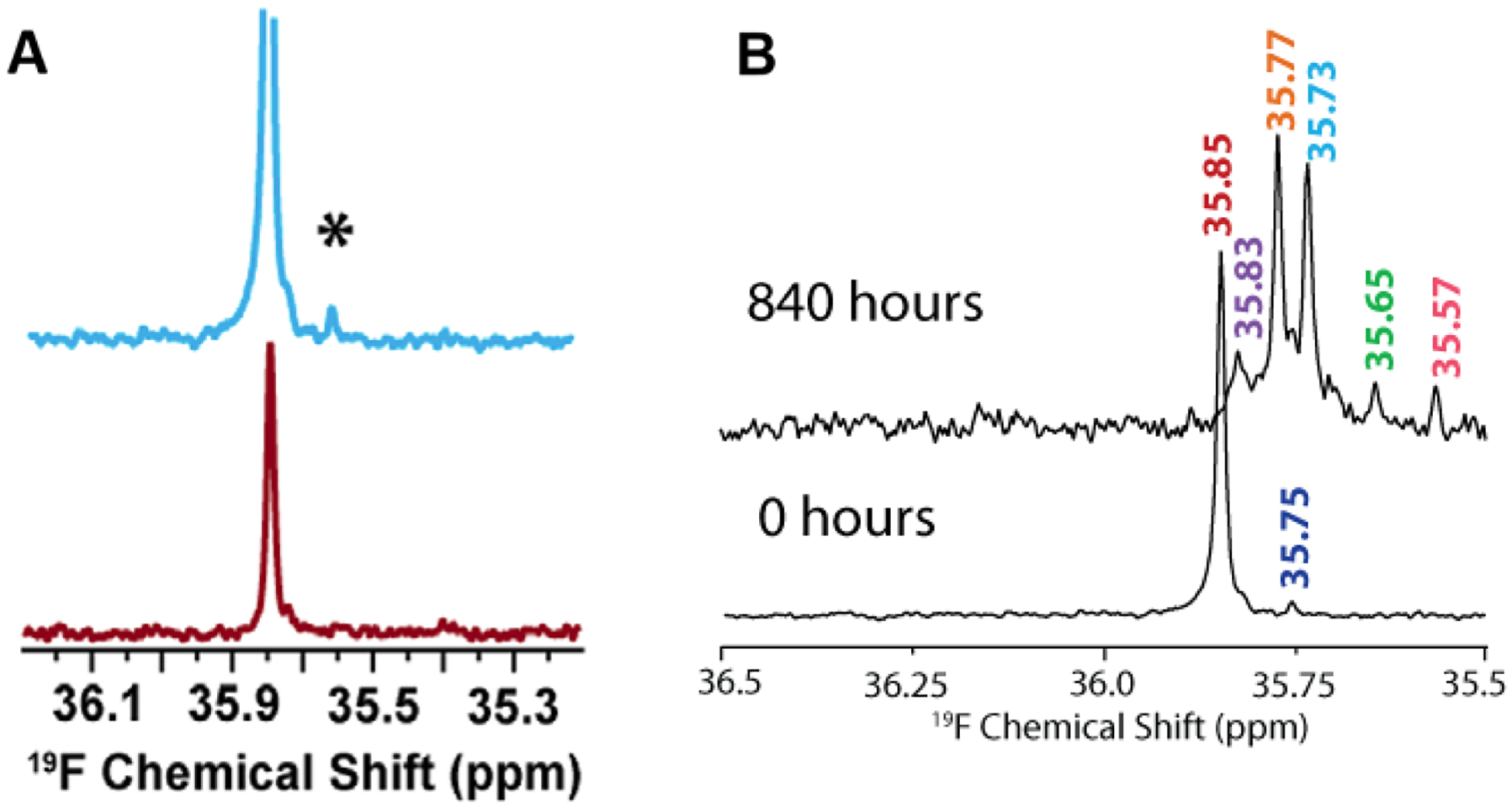
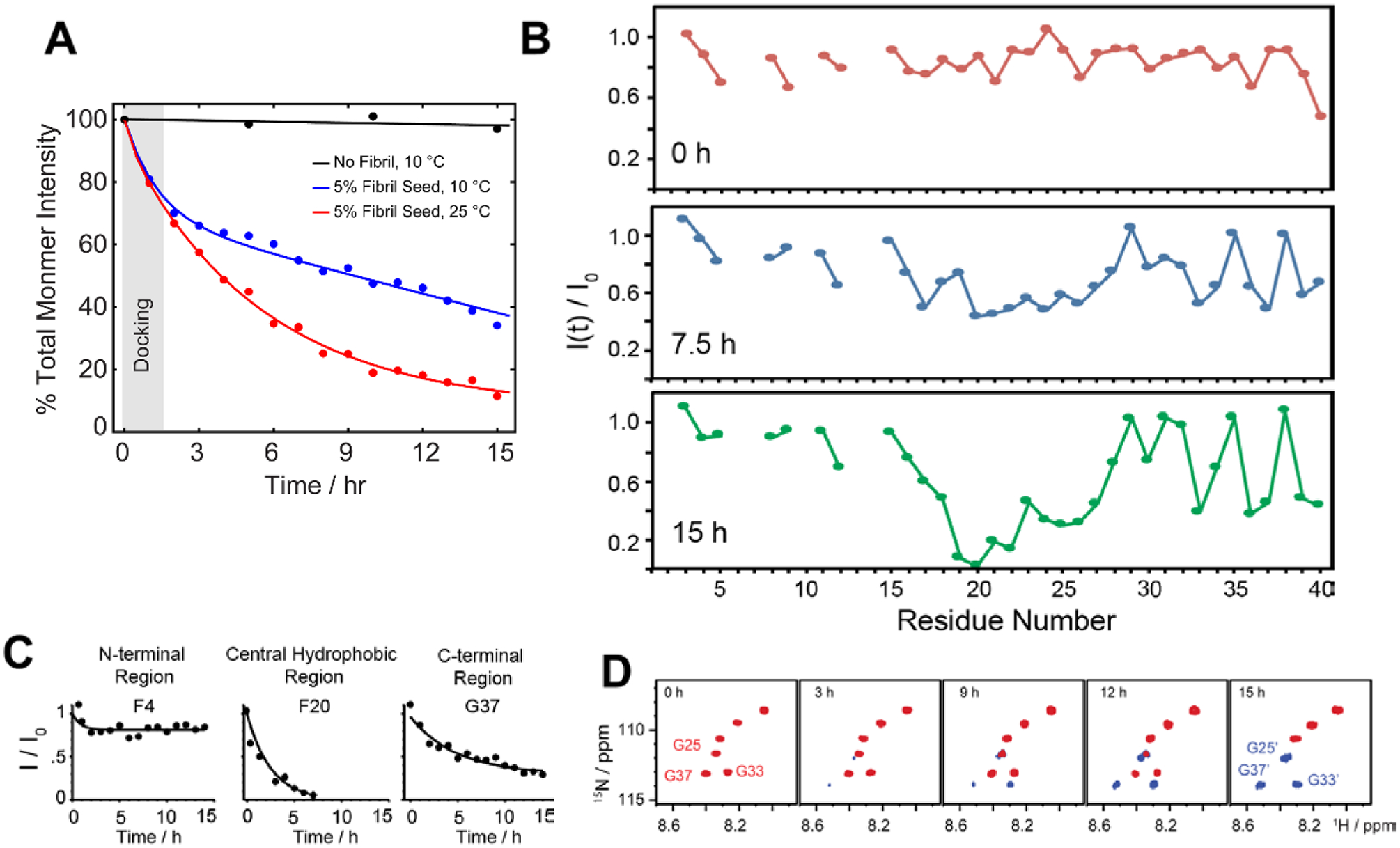
References
-
- Harrison RS, Sharpe PC, Singh Y and Fairlie DP, Rev. Physiol. Biochem. Pharmacol, 2007, 159, 1–77. - PubMed
-
- Chiti F and Dobson CM, Annu. Rev. Biochem, 2006, 75, 333–366. - PubMed
-
- Knowles TPJ, Vendruscolo M and Dobson CM, Nat. Rev. Mol. Cell Biol, 2014, 15, 384–396. - PubMed
-
- Hamley IW, Chem. Rev, 2012, 112, 5147–92. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources