

Biallelic mutations in WRAP53 result in dysfunctional telomeres, Cajal bodies and DNA repair, thereby causing Hoyeraal-Hreidarsson syndrome
- PMID: 32303682
- PMCID: PMC7165179
- DOI: 10.1038/s41419-020-2421-4
Biallelic mutations in WRAP53 result in dysfunctional telomeres, Cajal bodies and DNA repair, thereby causing Hoyeraal-Hreidarsson syndrome
Abstract
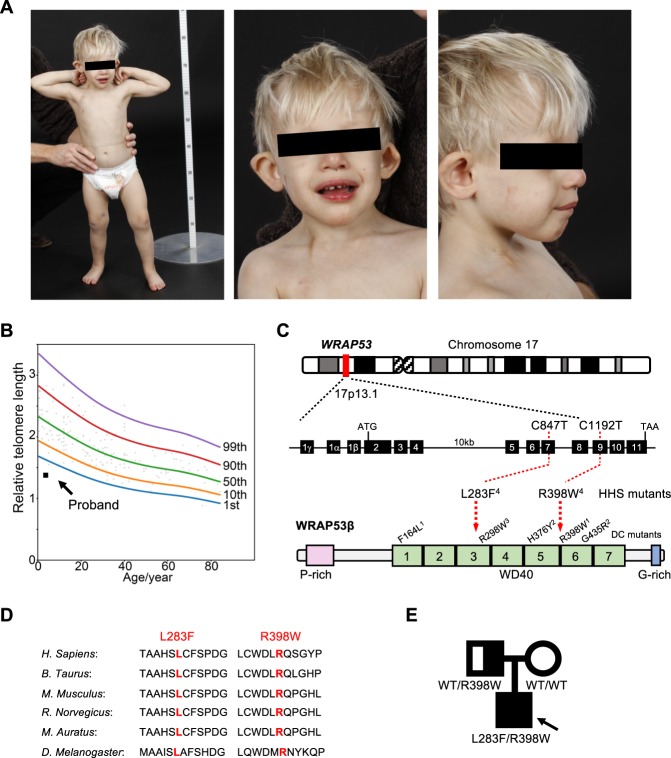
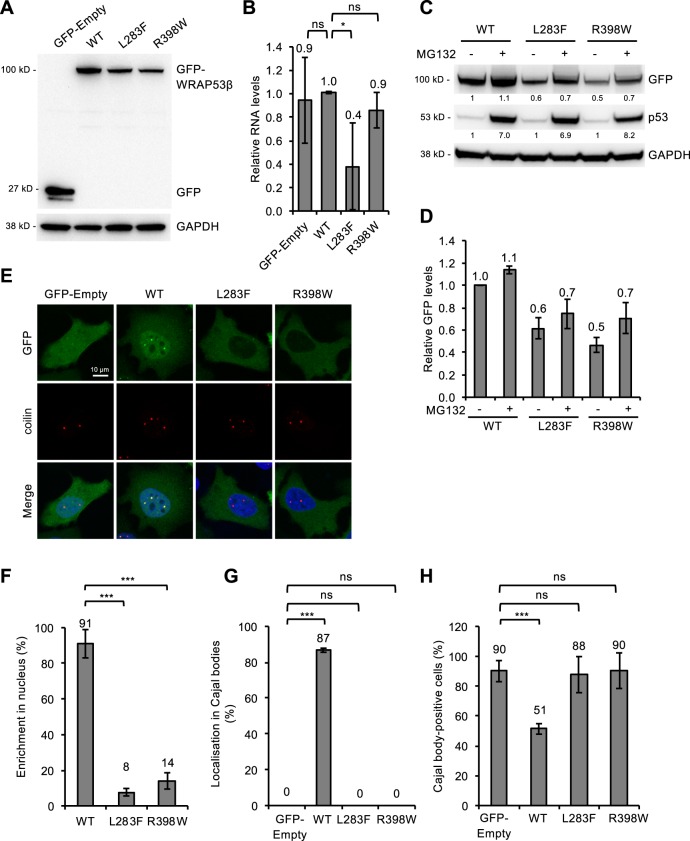
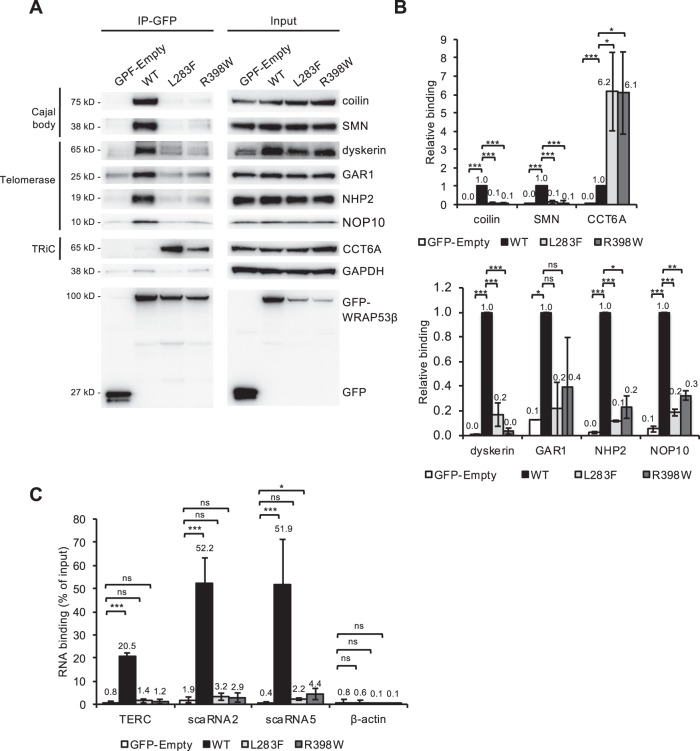
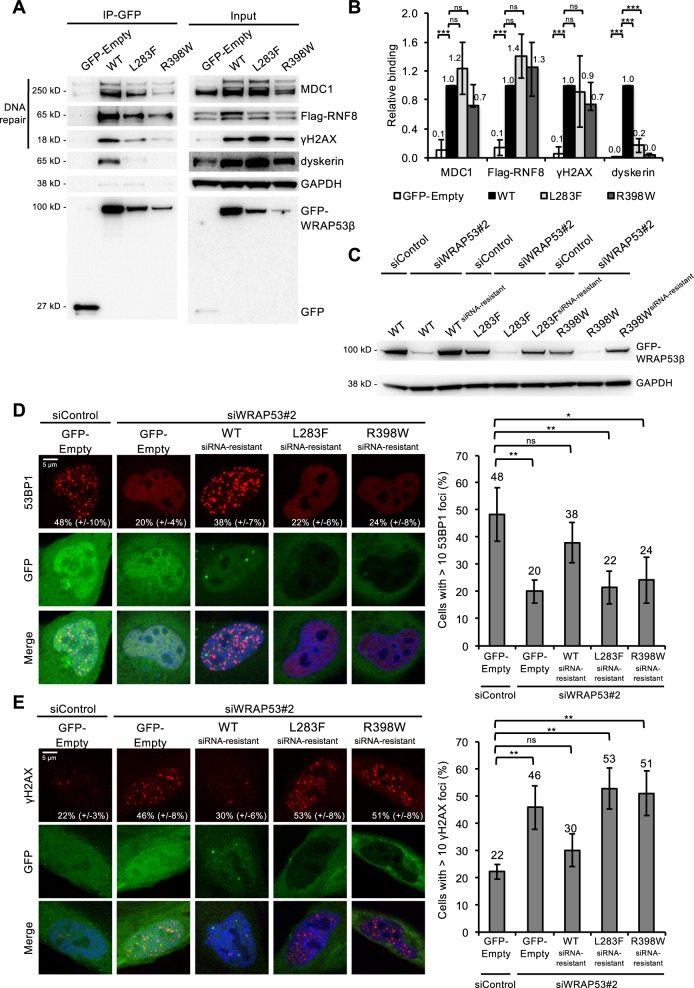
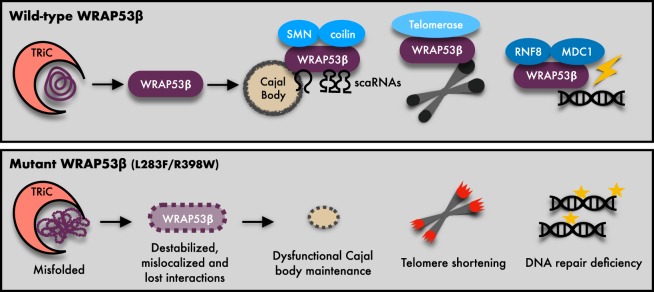
Approximately half of all cases of Hoyeraal-Hreidarsson syndrome (HHS), a multisystem disorder characterized by bone marrow failure, developmental defects and very short telomeres, are caused by germline mutations in genes related to telomere biology. However, the varying symptoms and severity of the disease indicate that additional mechanisms are involved. Here, a 3-year-old boy with HHS was found to carry biallelic germline mutations in WRAP53 (WD40 encoding RNA antisense to p53), that altered two highly conserved amino acids (L283F and R398W) in the WD40 scaffold domain of the protein encoded. WRAP53β (also known as TCAB1 or WDR79) is involved in intracellular trafficking of telomerase, Cajal body functions and DNA repair. We found that both mutations cause destabilization, mislocalization and faulty interactions of WRAP53β, defects linked to misfolding by the TRiC chaperonin complex. Consequently, WRAP53β HHS mutants cannot elongate telomeres, maintain Cajal bodies or repair DNA double-strand breaks. These findings provide a molecular explanation for the pathogenesis underlying WRAP53β-associated HHS and highlight the potential contribution of DNA damage and/or defects in Cajal bodies to the early onset and/or severity of this disease.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
