

Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma
- PMID: 32304665
- PMCID: PMC7266008
- DOI: 10.1016/j.cell.2020.03.062
Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma
Abstract
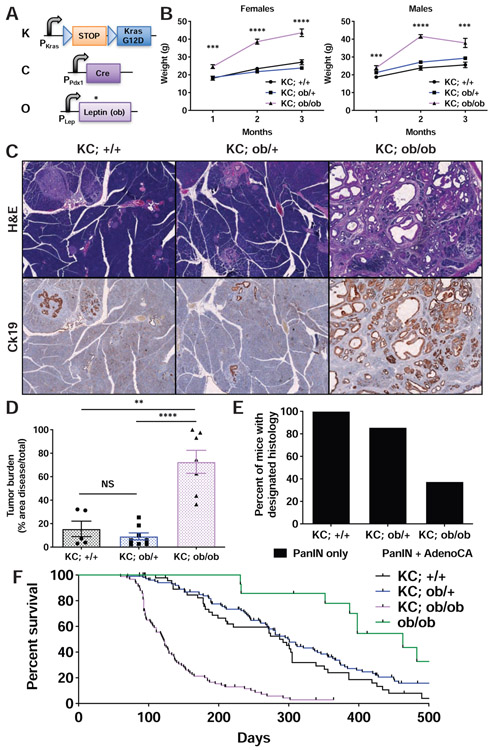
Obesity is a major modifiable risk factor for pancreatic ductal adenocarcinoma (PDAC), yet how and when obesity contributes to PDAC progression is not well understood. Leveraging an autochthonous mouse model, we demonstrate a causal and reversible role for obesity in early PDAC progression, showing that obesity markedly enhances tumorigenesis, while genetic or dietary induction of weight loss intercepts cancer development. Molecular analyses of human and murine samples define microenvironmental consequences of obesity that foster tumorigenesis rather than new driver gene mutations, including significant pancreatic islet cell adaptation in obesity-associated tumors. Specifically, we identify aberrant beta cell expression of the peptide hormone cholecystokinin (Cck) in response to obesity and show that islet Cck promotes oncogenic Kras-driven pancreatic ductal tumorigenesis. Our studies argue that PDAC progression is driven by local obesity-associated changes in the tumor microenvironment and implicate endocrine-exocrine signaling beyond insulin in PDAC development.
Keywords: beta cells; cholecystokinin; genetically engineered mouse models; leptin; obesity; pancreatic cancer; pancreatic islets; tumor microenvironment.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests K.J.D. is currently an employee of Sherlock Biosciences. M.D.B. acknowledges research support from ViaCyte and Dexcom and serves on the medical advisory boards for Novo Nordisk and ARIEL Precision Medicine. A.L.G. has received honoraria from Merck and Novo Nordisk and has received research funding from Novo Nordisk. M.I.M. has served on advisory panels for Pfizer, Novo Nordisk, and Zoe Global; he has received honoraria from Merck, Pfizer, Novo Nordisk, and Eli Lilly and research funding from Abbvie, Astra Zeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Merck, Novo Nordisk, Pfizer, Roche, Sanofi Aventis, Servier, and Takeda. As of June 2019, M.I.M. is an employee of Genentech and a holder of Roche stock. R.G.K. is co-founder and a Scientific Advisory Board member of Elucidata; a Consultant/Advisory Board member for Agios, Janssen, BI-Lilly, and Pfizer; and a recipient of sponsored research agreements from Agios, AstraZeneca/BMS, Lilly, Pfizer, and Poxel. S.K. is a paid scientific advisor to AI Therapeutics. B.M.W. declares research funding from Celgene and Eli Lilly & Company, and consults for BioLineRx, Celgene, G1 Therapeutics, and GRAIL. T.J. is a Board of Directors member of Amgen and Thermo Fisher Scientific, co-founder and Scientific Advisory Board member of Dragonfly Therapeutics, co-founder of T2 Biosystems, and Scientific Advisory Board member of SQZ Biotech with equity holding in all five companies; he receives funding from the Johnson & Johnson Lung Cancer Initiative and Calico. C.S.F. reports receiving personal fees from Eli Lilly, Entrinsic Health, Pfizer, Merck, Sanofi, Roche, Genentech, Merrimack Pharma, Dicerna, Bayer, Celgene, Agios, Gilead Sciences, Five Prime Therapeutics, Taiho, KEW, and CytomX Therapeutics and receiving support from CytomX Therapeutics. M.D.M. acknowledges research support from Genentech.
Figures






Comment in
-
Not the usual suspect.Nat Rev Cancer. 2020 Jun;20(6):302. doi: 10.1038/s41568-020-0271-0. Nat Rev Cancer. 2020. PMID: 32376857 No abstract available.
-
Endocrine-exocrine signals in obesity-associated pancreatic cancer.Nat Rev Gastroenterol Hepatol. 2020 Aug;17(8):455-456. doi: 10.1038/s41575-020-0324-6. Nat Rev Gastroenterol Hepatol. 2020. PMID: 32483354 Free PMC article.
References
-
- Babic A, Bao Y, Qian ZR, Yuan C, Giovannucci EL, Aschard H, Kraft P, Amundadottir LT, Stolzenberg-Solomon R, Morales-Oyarvide V, et al. (2016). Pancreatic Cancer Risk Associated with Prediagnostic Plasma Levels of Leptin and Leptin Receptor Genetic Polymorphisms. Cancer Res 76, 7160–7167. - PMC - PubMed
-
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. (2016). Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52. - PubMed
Publication types
MeSH terms
Grants and funding
- 098381/WT_/Wellcome Trust/United Kingdom
- P30 CA016672/CA/NCI NIH HHS/United States
- U01 CA210171/CA/NCI NIH HHS/United States
- U01 DK085545/DK/NIDDK NIH HHS/United States
- 203141/WT_/Wellcome Trust/United Kingdom
- U01 DK105535/DK/NIDDK NIH HHS/United States
- KL2 TR001999/TR/NCATS NIH HHS/United States
- P30 DK045735/DK/NIDDK NIH HHS/United States
- F31 HD097958/HD/NICHD NIH HHS/United States
- 212259/WT_/Wellcome Trust/United Kingdom
- 090532/WT_/Wellcome Trust/United Kingdom
- P30 CA016359/CA/NCI NIH HHS/United States
- L30 CA171131/CA/NCI NIH HHS/United States
- 200837/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- K07 CA222159/CA/NCI NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- K08 CA208016/CA/NCI NIH HHS/United States
- P01 CA042063/CA/NCI NIH HHS/United States
- R01 GM130847/GM/NIGMS NIH HHS/United States
- S10 OD018521/OD/NIH HHS/United States
- P30 CA014051/CA/NCI NIH HHS/United States
- DP2 CA248136/CA/NCI NIH HHS/United States
- 200837/WT_/Wellcome Trust/United Kingdom
- 106130/WT_/Wellcome Trust/United Kingdom
- MR/L020149/1/MRC_/Medical Research Council/United Kingdom
- R01 DK110181/DK/NIDDK NIH HHS/United States
- 095101/WT_/Wellcome Trust/United Kingdom
- T32 GM007499/GM/NIGMS NIH HHS/United States
- U19 AI089992/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
