

Supporting pandemic response using genomics and bioinformatics: A case study on the emergent SARS-CoV-2 outbreak
- PMID: 32306500
- PMCID: PMC7264654
- DOI: 10.1111/tbed.13588
Supporting pandemic response using genomics and bioinformatics: A case study on the emergent SARS-CoV-2 outbreak
Abstract
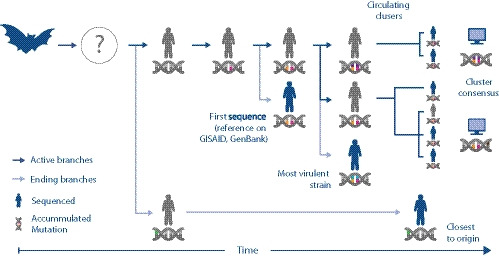
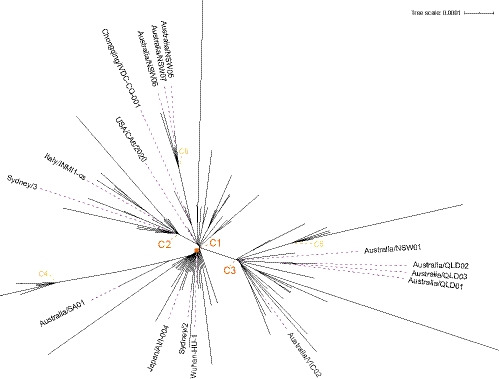
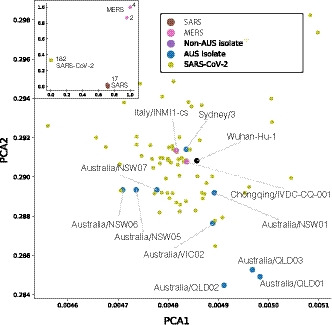
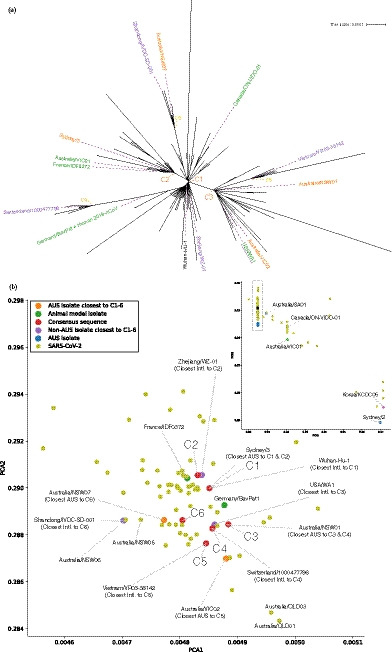
Pre-clinical responses to fast-moving infectious disease outbreaks heavily depend on choosing the best isolates for animal models that inform diagnostics, vaccines and treatments. Current approaches are driven by practical considerations (e.g. first available virus isolate) rather than a detailed analysis of the characteristics of the virus strain chosen, which can lead to animal models that are not representative of the circulating or emerging clusters. Here, we suggest a combination of epidemiological, experimental and bioinformatic considerations when choosing virus strains for animal model generation. We discuss the currently chosen SARS-CoV-2 strains for international coronavirus disease (COVID-19) models in the context of their phylogeny as well as in a novel alignment-free bioinformatic approach. Unlike phylogenetic trees, which focus on individual shared mutations, this new approach assesses genome-wide co-developing functionalities and hence offers a more fluid view of the 'cloud of variances' that RNA viruses are prone to accumulate. This joint approach concludes that while the current animal models cover the existing viral strains adequately, there is substantial evolutionary activity that is likely not considered by the current models. Based on insights from the non-discrete alignment-free approach and experimental observations, we suggest isolates for future animal models.
Keywords: COVID-19; PHEIC; alignment-free phylogeny; bioinformatics; genomics; viral evolution.
© 2020 The Authors. Transboundary and Emerging Diseases published by Blackwell Verlag GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- CSIRO (2020). Working against the new coronavirus. March 5, 2020, Retrieved from https://www.csiro.au/en/Research/Health/Infectious‐dieases‐coronavirus/c...
-
- Eigen, M. , McCaskill, J. , & Schuster, P. (1988). Molecular quasi‐species. The Journal of Physical Chemistry, 92(24), 6881–6891. 10.1021/j100335a010 - DOI
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
