

Targeting Rho GTPase Signaling Networks in Cancer
- PMID: 32309283
- PMCID: PMC7145979
- DOI: 10.3389/fcell.2020.00222
Targeting Rho GTPase Signaling Networks in Cancer
Abstract
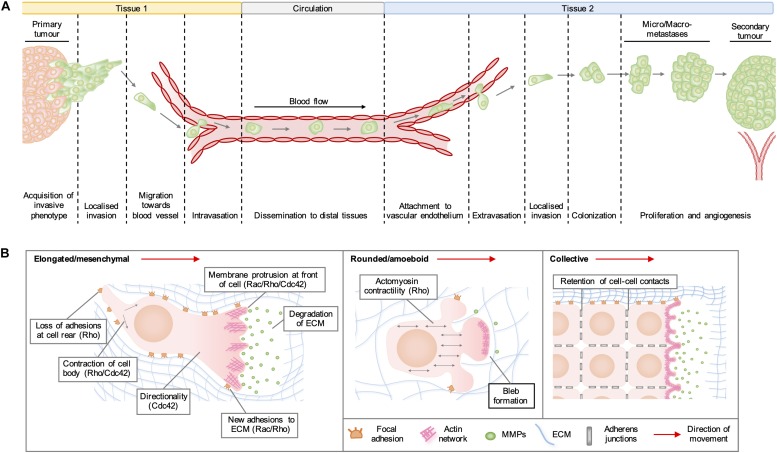
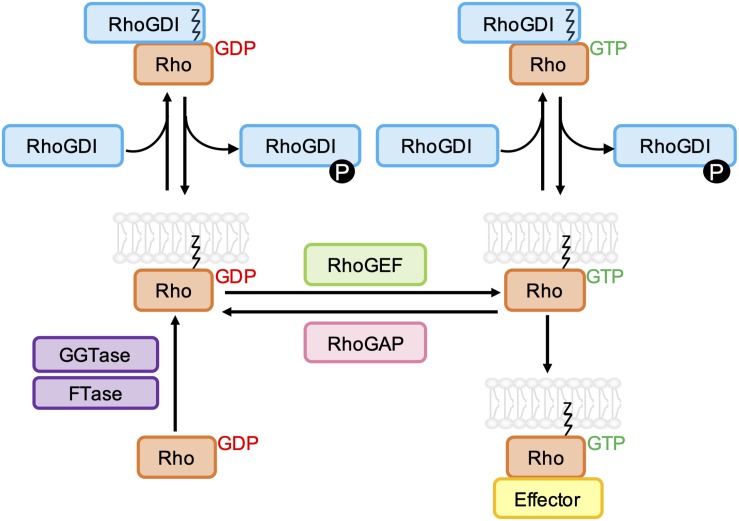
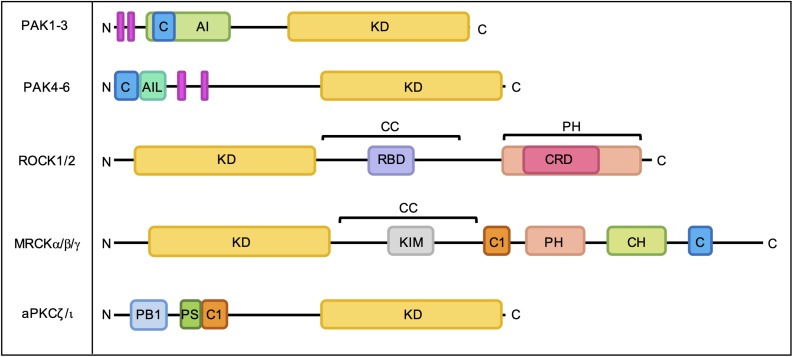
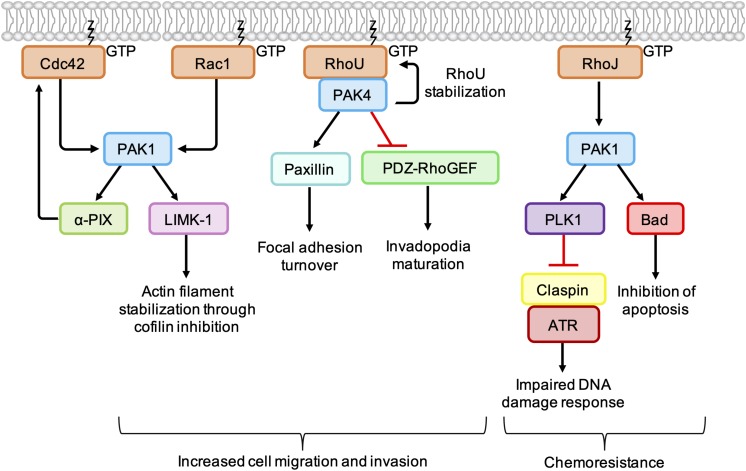
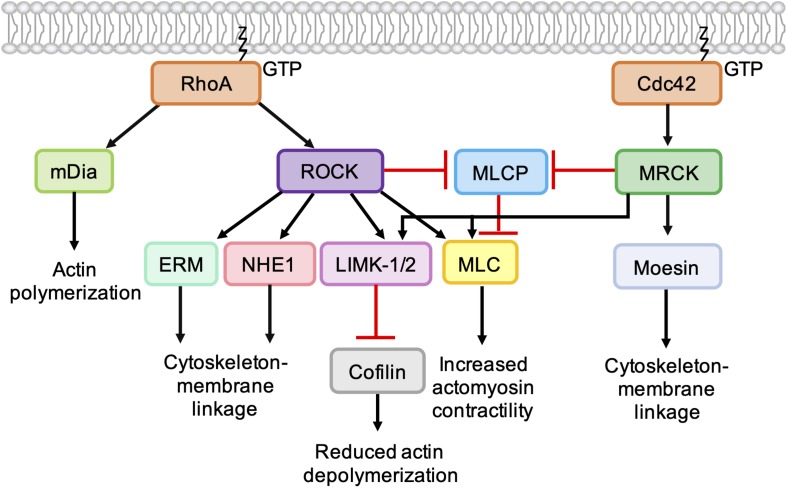
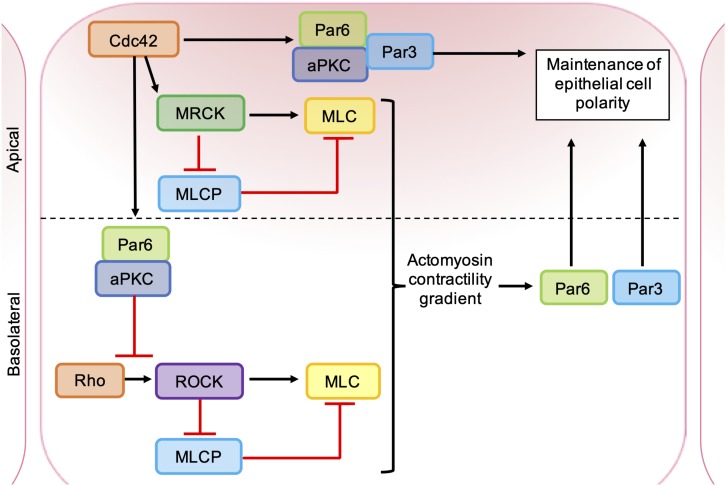
As key regulators of cytoskeletal dynamics, Rho GTPases coordinate a wide range of cellular processes, including cell polarity, cell migration, and cell cycle progression. The adoption of a pro-migratory phenotype enables cancer cells to invade the stroma surrounding the primary tumor and move toward and enter blood or lymphatic vessels. Targeting these early events could reduce the progression to metastatic disease, the leading cause of cancer-related deaths. Rho GTPases play a key role in the formation of dynamic actin-rich membrane protrusions and the turnover of cell-cell and cell-extracellular matrix adhesions required for efficient cancer cell invasion. Here, we discuss the roles of Rho GTPases in cancer, their validation as therapeutic targets and the challenges of developing clinically viable Rho GTPase inhibitors. We review other therapeutic targets in the wider Rho GTPase signaling network and focus on the four best characterized effector families: p21-activated kinases (PAKs), Rho-associated protein kinases (ROCKs), atypical protein kinase Cs (aPKCs), and myotonic dystrophy kinase-related Cdc42-binding kinases (MRCKs).
Keywords: GTPase; Rho; cancer; invasion; metastasis.
Copyright © 2020 Clayton and Ridley.
Figures
References
-
- Aguilar B. J., Zhao Y., Zhou H., Huo S., Chen Y. H., Lu Q. (2019). Inhibition of Cdc42-intersectin interaction by small molecule ZCL367 impedes cancer cell cycle progression, proliferation, migration, and tumor growth. Cancer Biol. Ther. 20 740–749. 10.1080/15384047.2018.1564559 - DOI - PMC - PubMed
-
- Amano M., Fukata Y., Shimokawa H., Kaibuchi K. (2000). Purification and in vitro activity of Rho-associated kinase. Methods Enzymol. 325 149–155. - PubMed
-
- Apostolatos A. H., Ratnayake W. S., Win-Piazza H., Apostolatos C. A., Smalley T., Kang L., et al. (2018). Inhibition of atypical protein kinase Ciota effectively reduces the malignancy of prostate cancer cells by downregulating the NF-kappaB signaling cascade. Int. J. Oncol. 53 1836–1846. 10.3892/ijo.2018.4542 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous