

Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment
- PMID: 32312953
- PMCID: PMC7170895
- DOI: 10.1038/s41419-020-2449-5
Enhanced fatty acid oxidation provides glioblastoma cells metabolic plasticity to accommodate to its dynamic nutrient microenvironment
Abstract
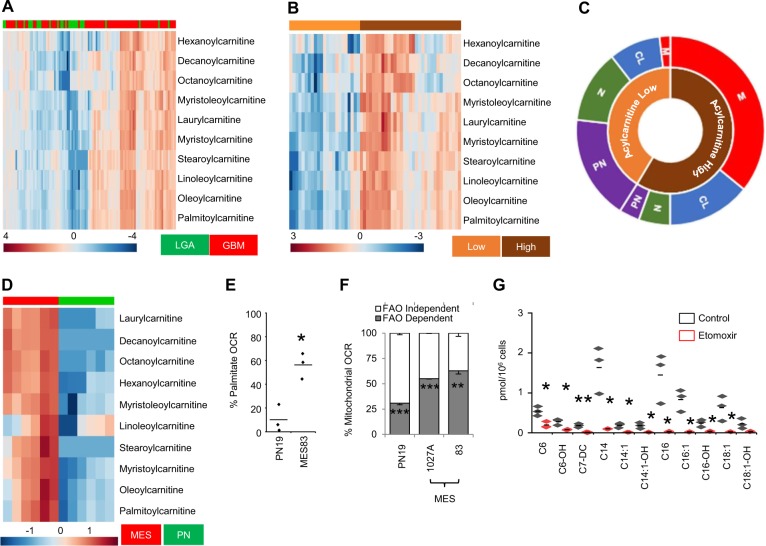
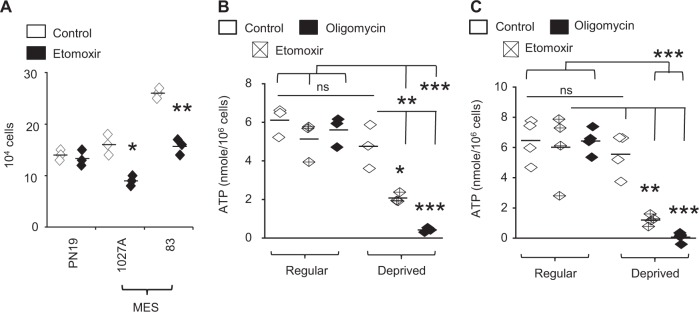
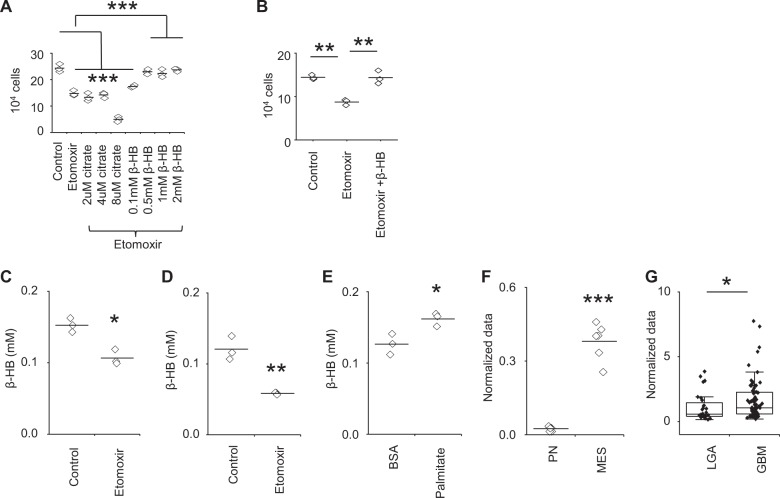
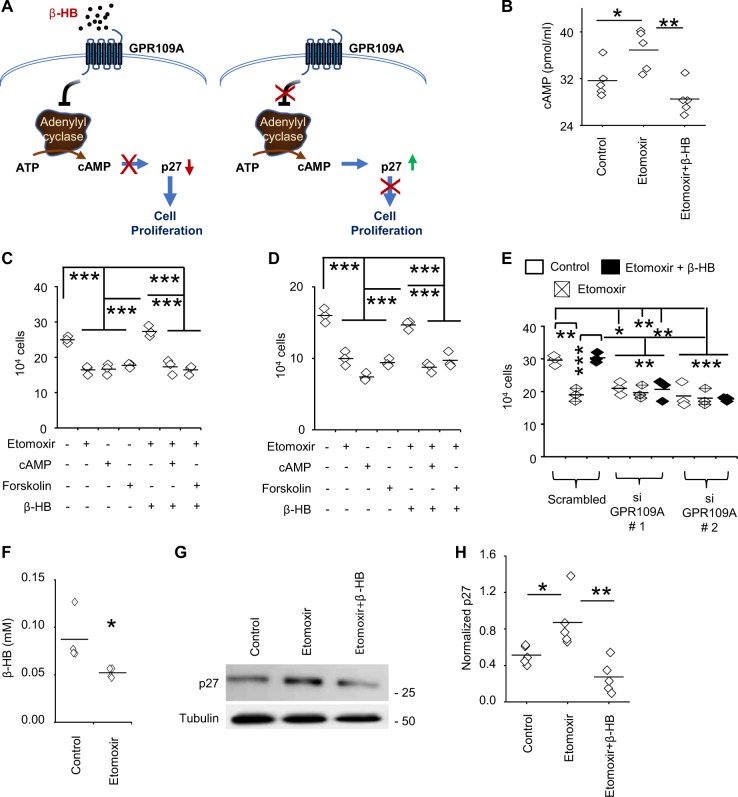
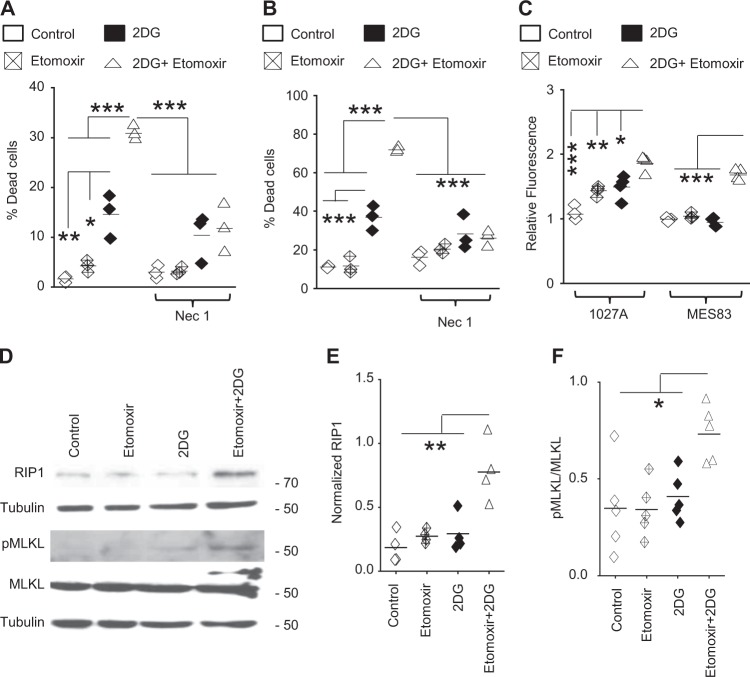
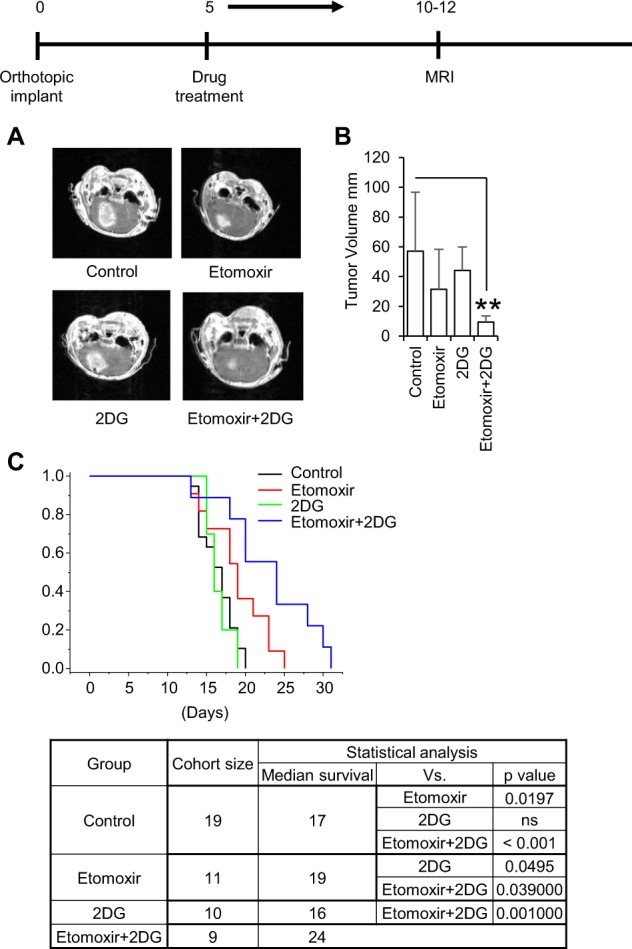
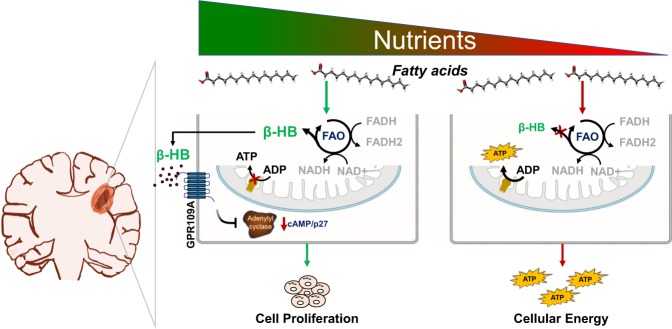
Despite advances in molecularly characterizing glioblastoma (GBM), metabolic alterations driving its aggressive phenotype are only beginning to be recognized. Integrative cross-platform analysis coupling global metabolomic and gene expression profiling on patient-derived glioma identified fatty acid β-oxidation (FAO) as a metabolic node in GBM. We determined that the biologic consequence of enhanced FAO is directly dependent upon tumor microenvironment. FAO serves as a metabolic cue to drive proliferation in a β-HB/GPR109A dependent autocrine manner in nutrient favorable conditions, while providing an efficient, alternate source of ATP only in nutrient unfavorable conditions. Rational combinatorial strategies designed to target these dynamic roles FAO plays in gliomagenesis resulted in necroptosis-mediated metabolic synthetic lethality in GBM. In summary, we identified FAO as a dominant metabolic node in GBM that provides metabolic plasticity, allowing these cells to adapt to their dynamic microenvironment. Combinatorial strategies designed to target these diverse roles FAO plays in gliomagenesis offers therapeutic potential in GBM.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
