

Perturbing proteomes at single residue resolution using base editing
- PMID: 32313011
- PMCID: PMC7170841
- DOI: 10.1038/s41467-020-15796-7
Perturbing proteomes at single residue resolution using base editing
Abstract
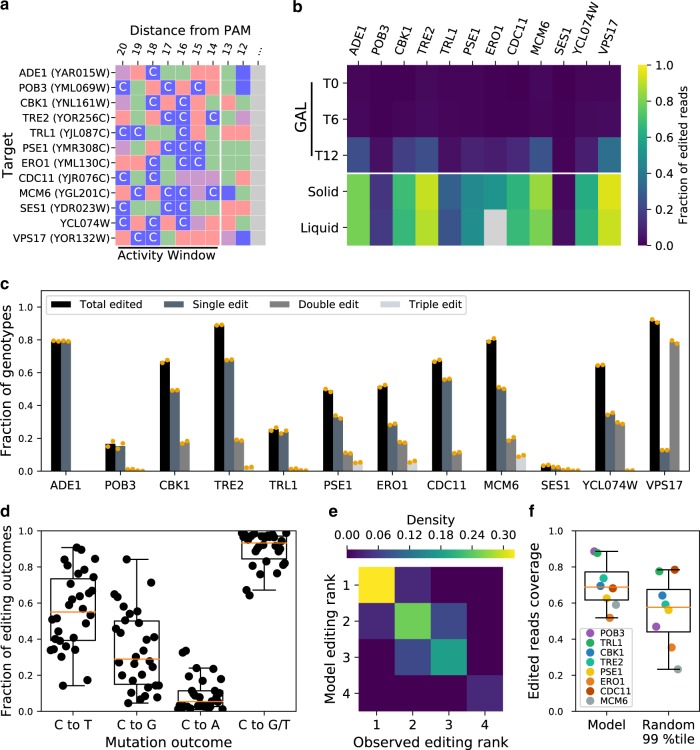
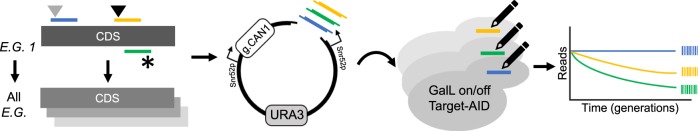
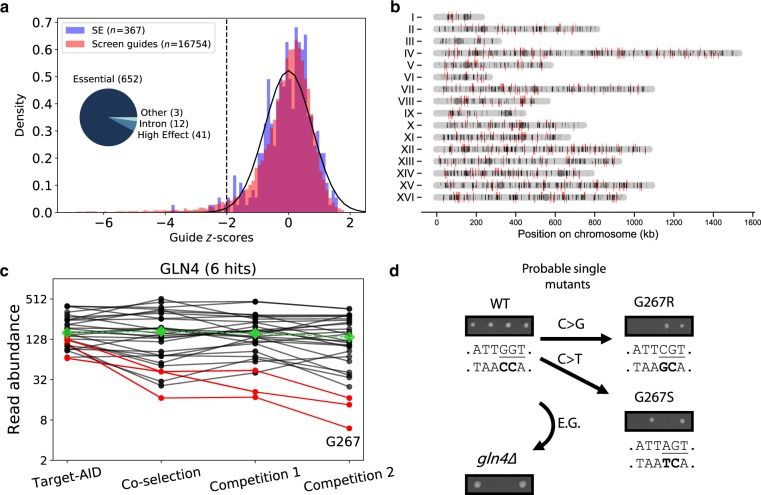
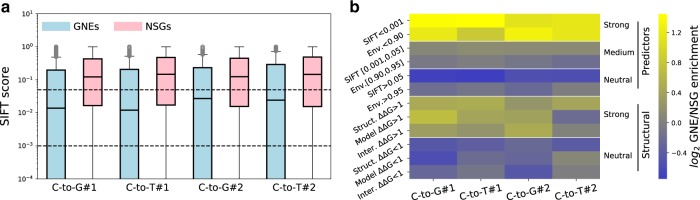
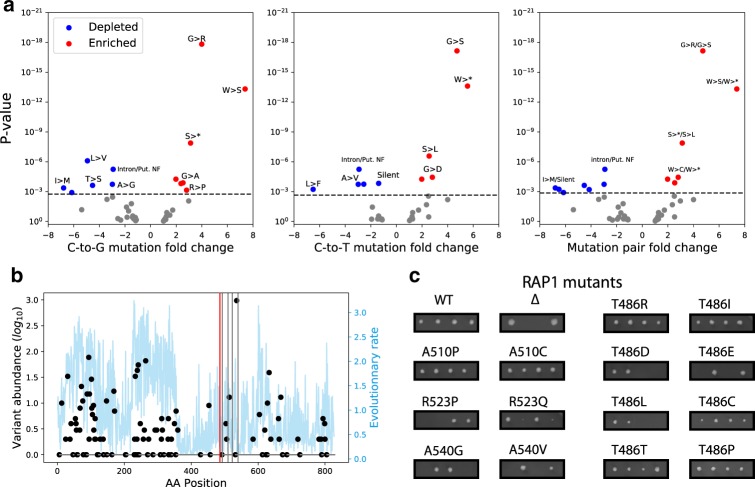
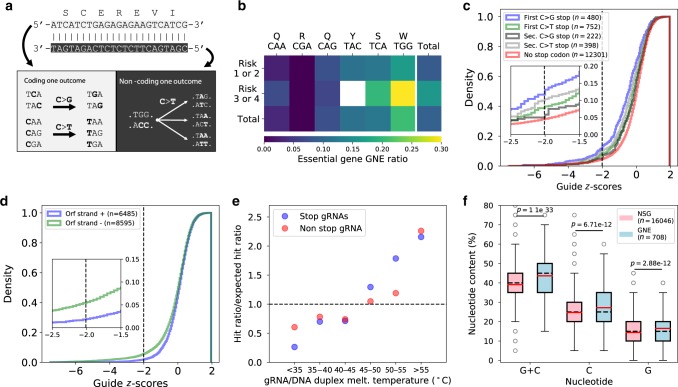
Base editors derived from CRISPR-Cas9 systems and DNA editing enzymes offer an unprecedented opportunity for the precise modification of genes, but have yet to be used at a genome-scale throughput. Here, we test the ability of the Target-AID base editor to systematically modify genes genome-wide by targeting yeast essential genes. We mutate around 17,000 individual sites in parallel across more than 1500 genes. We identify over 700 sites at which mutations have a significant impact on fitness. Using previously determined and preferred Target-AID mutational outcomes, we find that gRNAs with significant effects on fitness are enriched in variants predicted to be deleterious based on residue conservation and predicted protein destabilization. We identify key features influencing effective gRNAs in the context of base editing. Our results show that base editing is a powerful tool to identify key amino acid residues at the scale of proteomes.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
