

Liability threshold modeling of case-control status and family history of disease increases association power
- PMID: 32313248
- PMCID: PMC7210076
- DOI: 10.1038/s41588-020-0613-6
Liability threshold modeling of case-control status and family history of disease increases association power
Abstract
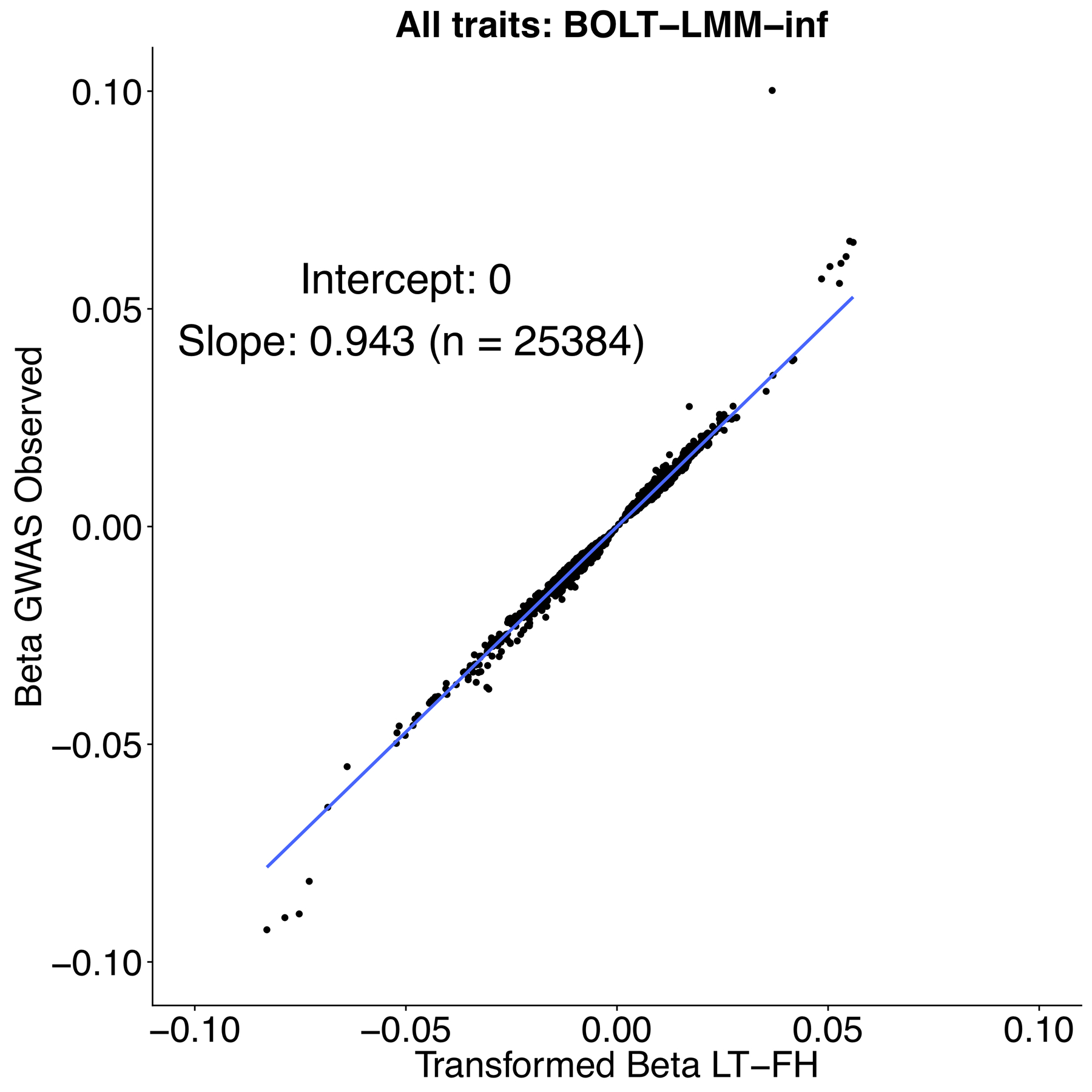
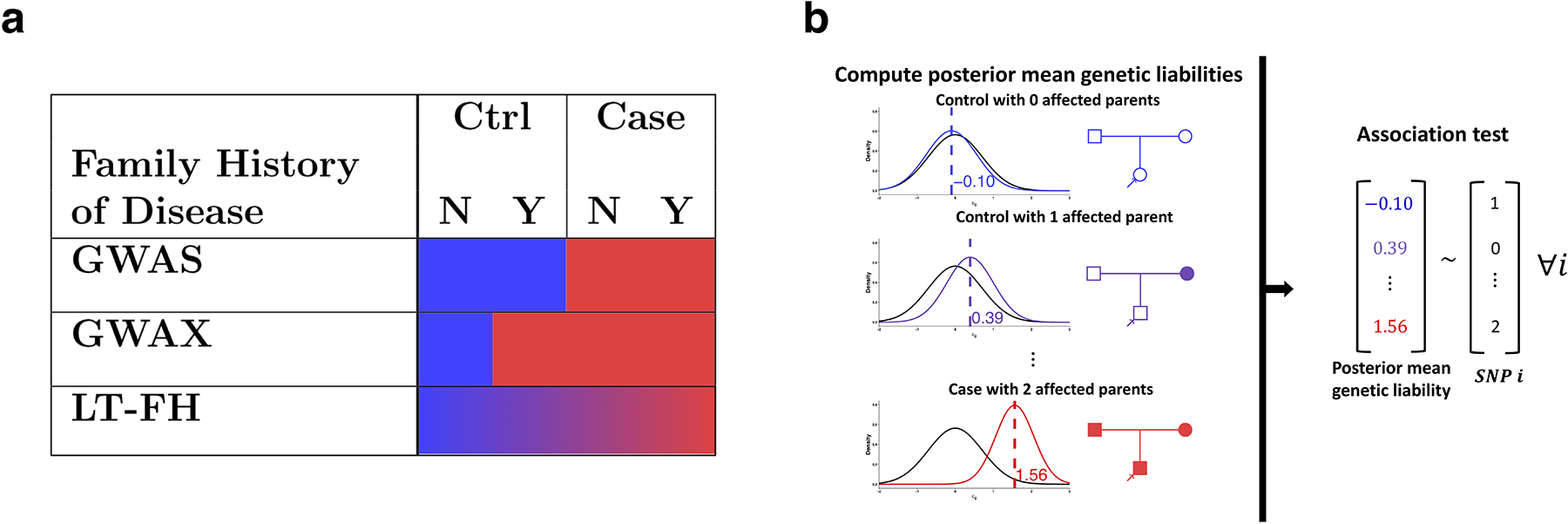
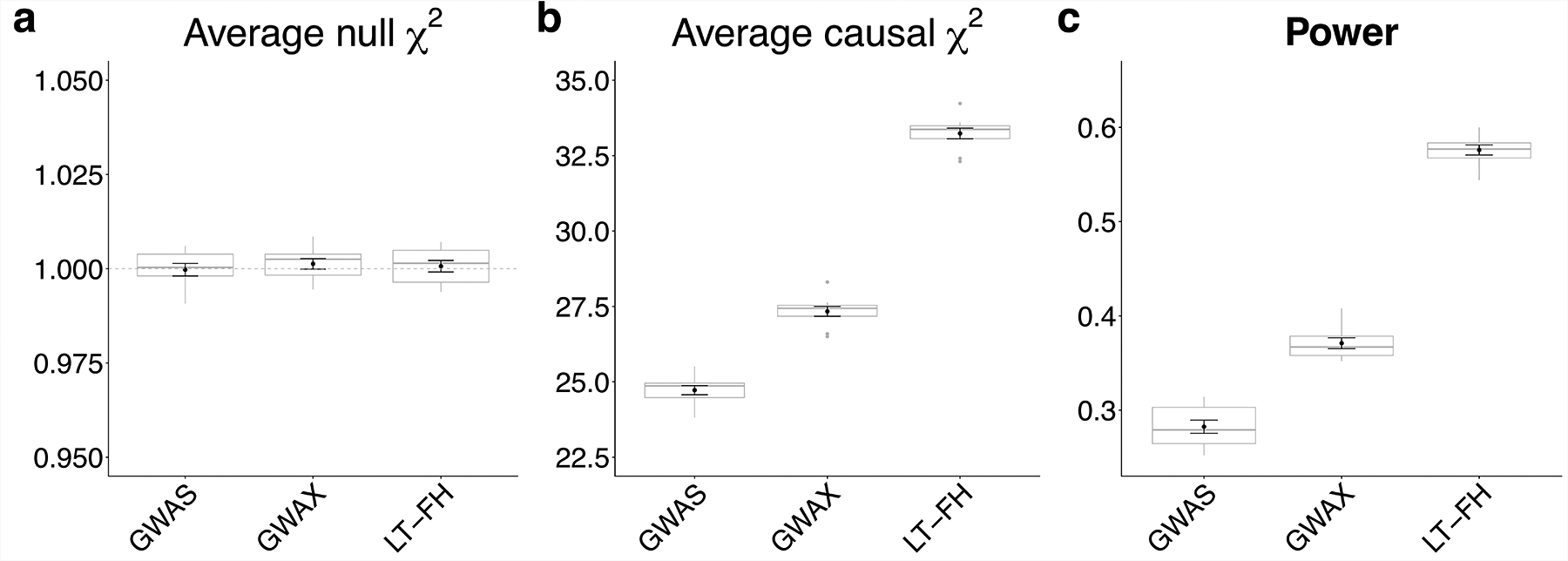
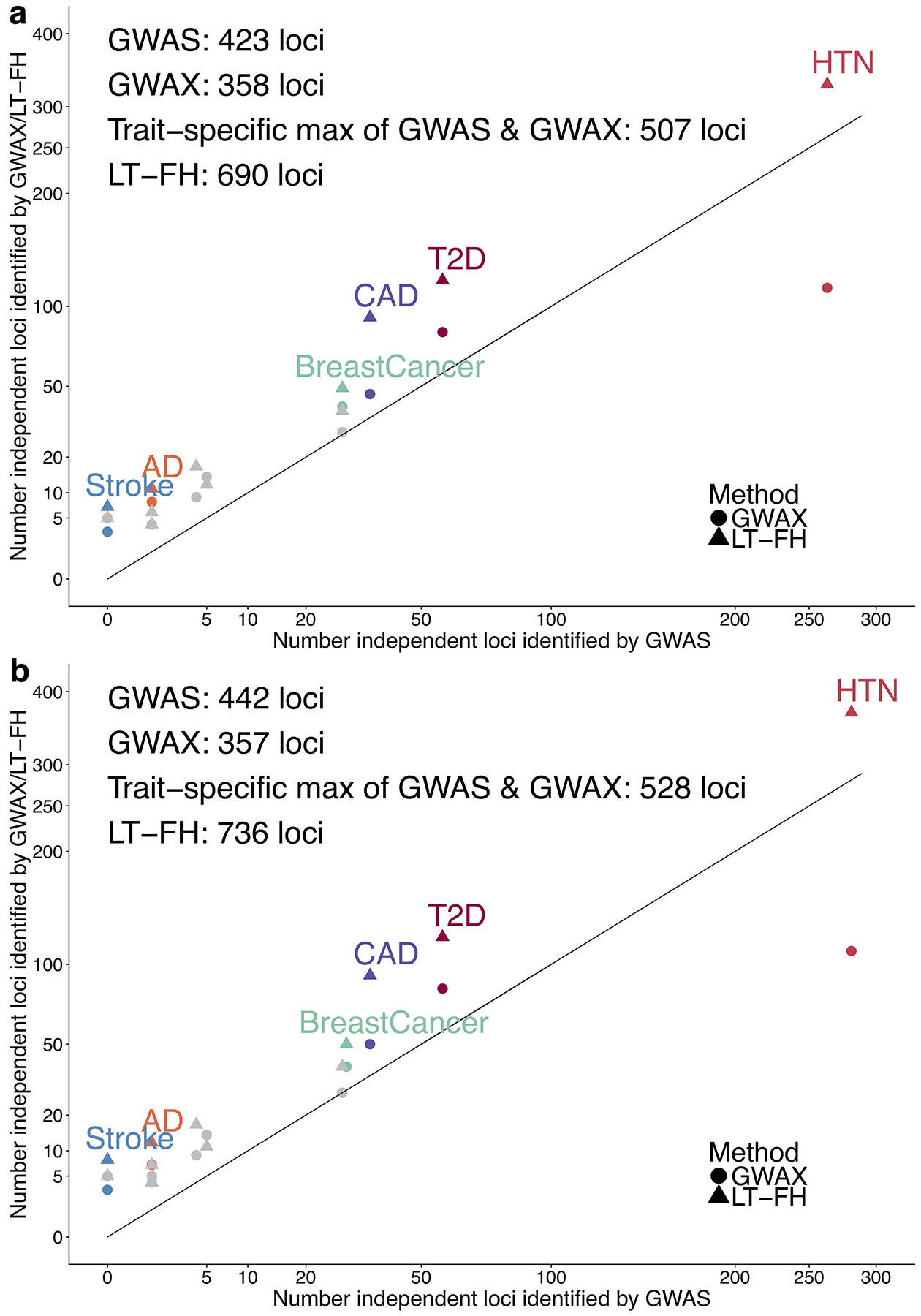
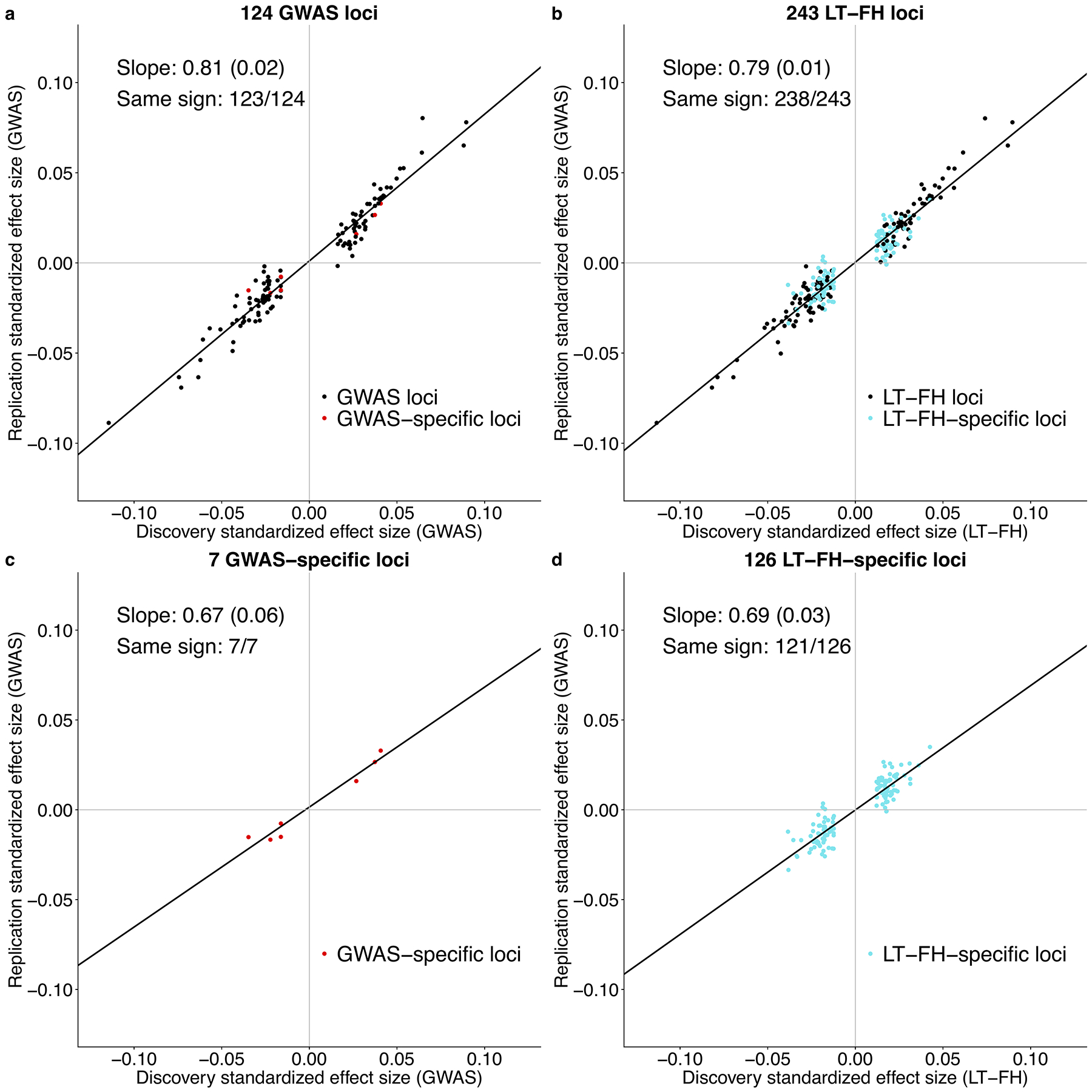
Family history of disease can provide valuable information in case-control association studies, but it is currently unclear how to best combine case-control status and family history of disease. We developed an association method based on posterior mean genetic liabilities under a liability threshold model, conditional on case-control status and family history (LT-FH). Analyzing 12 diseases from the UK Biobank (average N = 350,000) we compared LT-FH to genome-wide association without using family history (GWAS) and a previous proxy-based method incorporating family history (GWAX). LT-FH was 63% (standard error (s.e.) 6%) more powerful than GWAS and 36% (s.e. 4%) more powerful than the trait-specific maximum of GWAS and GWAX, based on the number of independent genome-wide-significant loci across all diseases (for example, 690 loci for LT-FH versus 423 for GWAS); relative improvements were similar when applying BOLT-LMM to GWAS, GWAX and LT-FH phenotypes. Thus, LT-FH greatly increases association power when family history of disease is available.
Conflict of interest statement
Competing Interests Statement
The authors declare no competing interests.
Figures
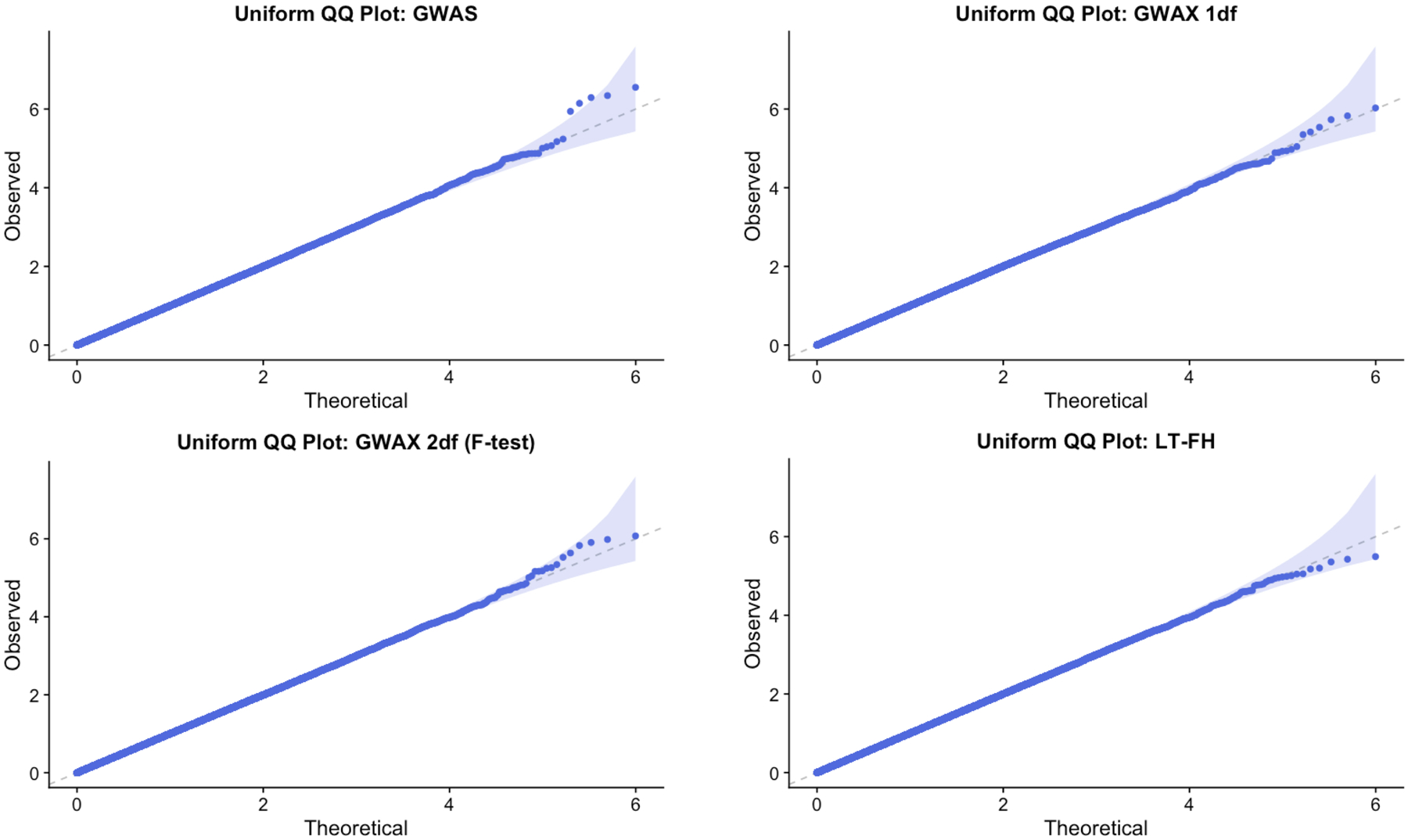
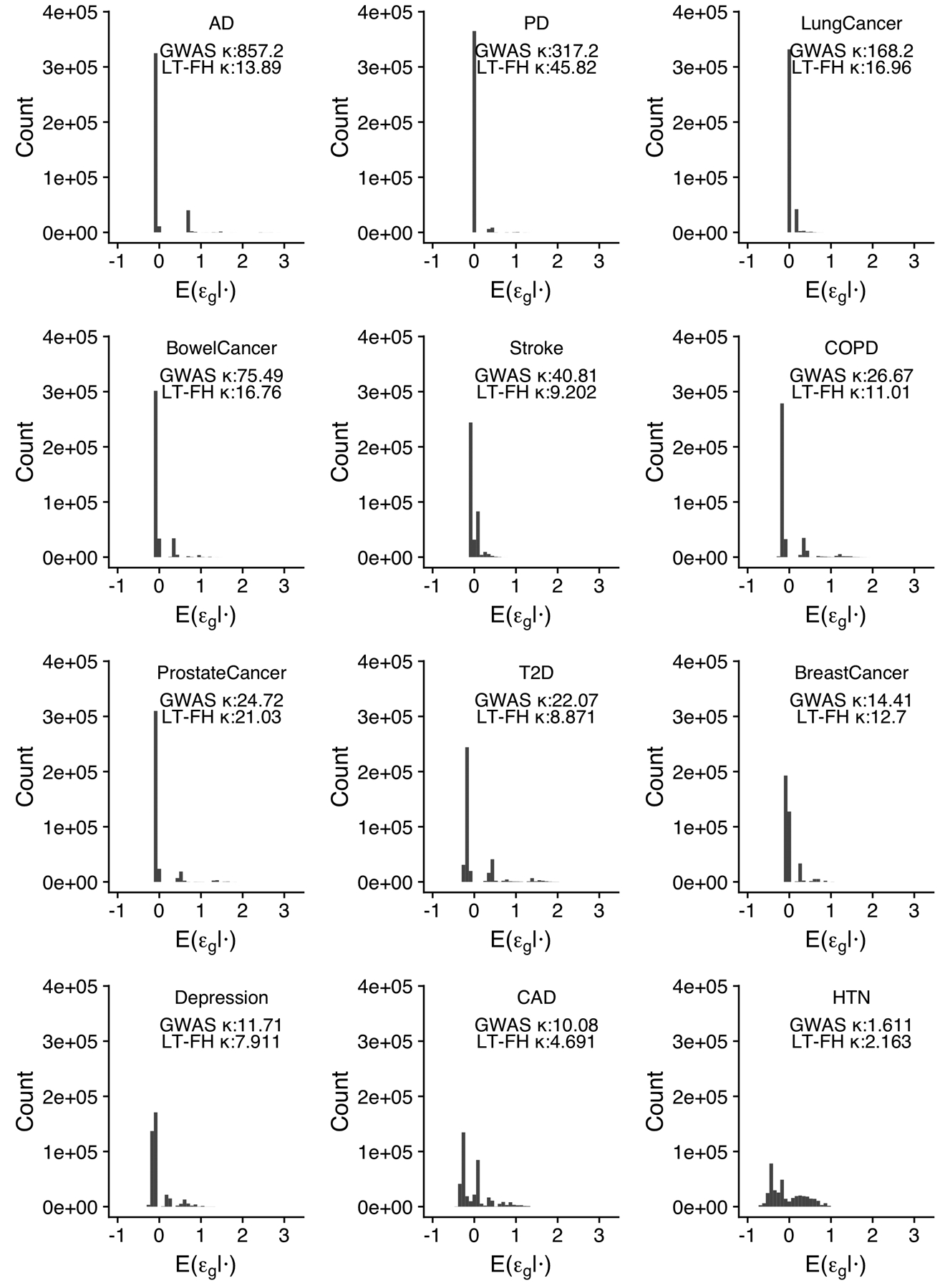
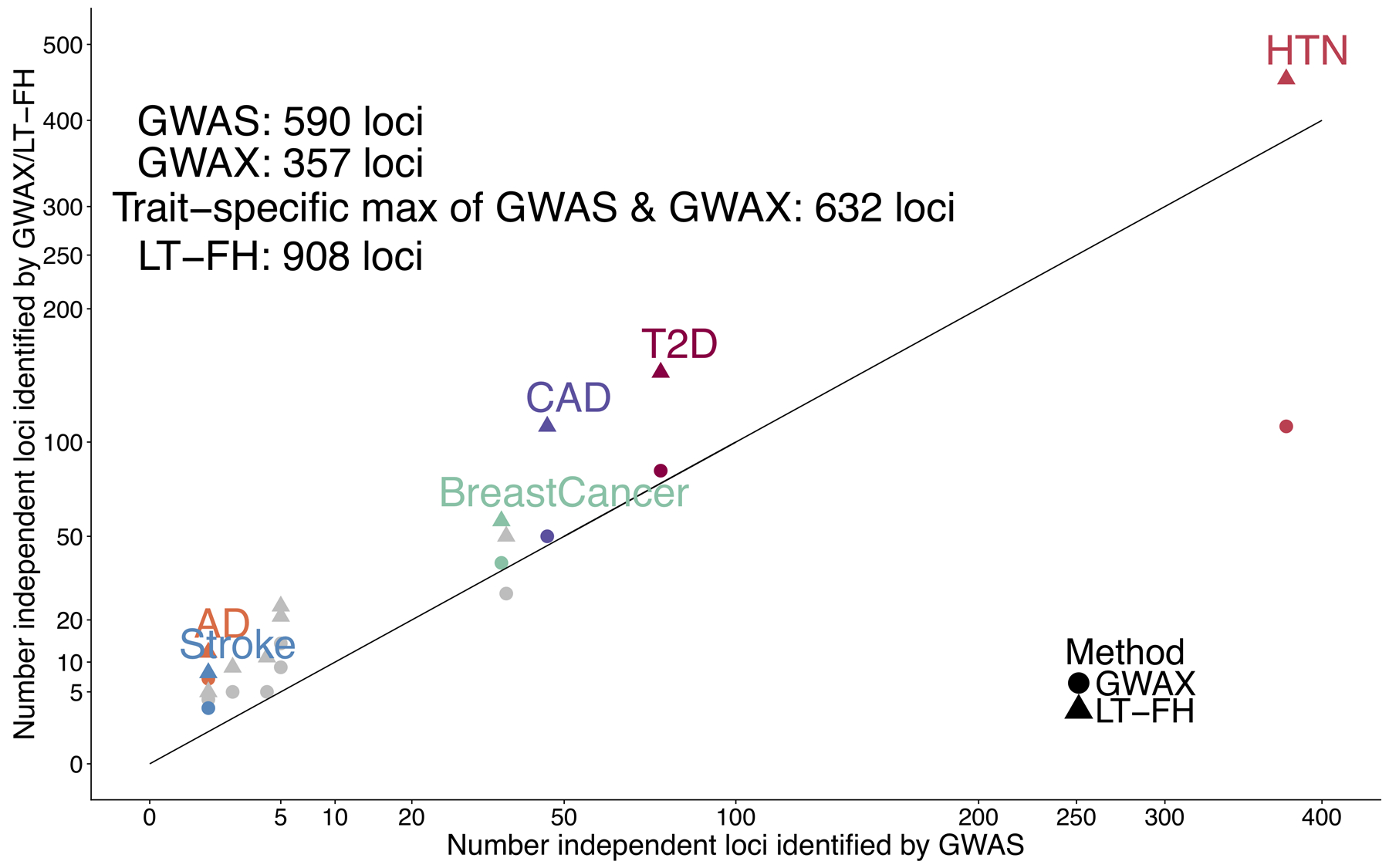
References
-
- Liu JZ, Erlich Y & Pickrell JK Case-control association mapping by proxy using family history of disease. Nat. Genet 49, 325–331 (2017). - PubMed
-
- Visscher PM & Duffy DL The Value of Relatives With Phenotypes But Missing Genotypes in Association Studies for Quantitative Traits. Genet. Epidemiol 30, 30–36 (2006). - PubMed
-
- Hayes BJ, Bowman PJ, Chamberlain AJ & Goddard ME Genomic selection in dairy cattle: Progress and challenges. J. Dairy Sci 92, 433–443 (2008). - PubMed
-
- Misztal I, Legarra A & Aguilar I Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information. J. Dairy Sci 92, 4648–4655 (2009). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
