

Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study
- PMID: 32315171
- PMCID: PMC7197972
- DOI: 10.1021/acs.jcim.0c00179
Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study
Abstract
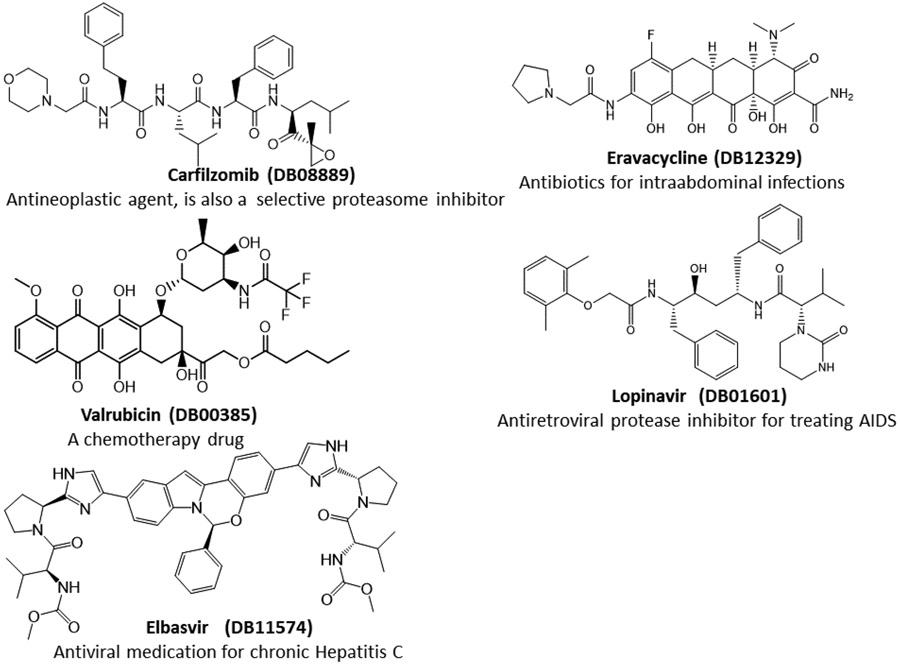
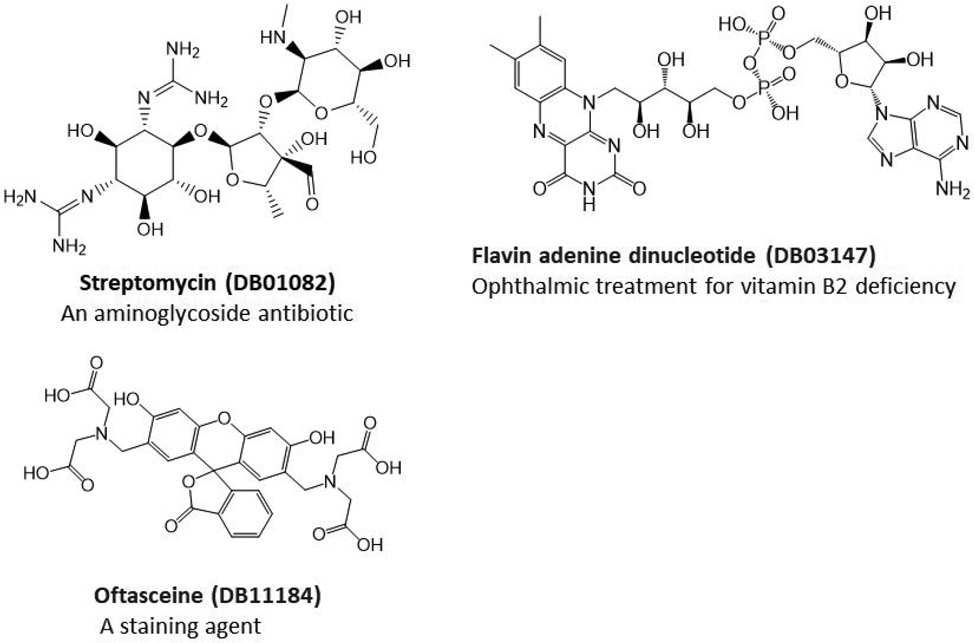
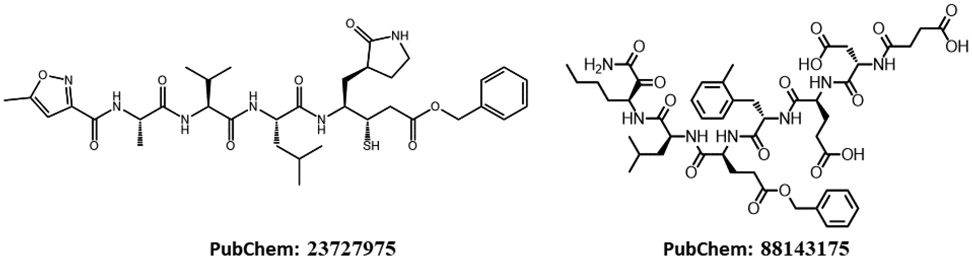
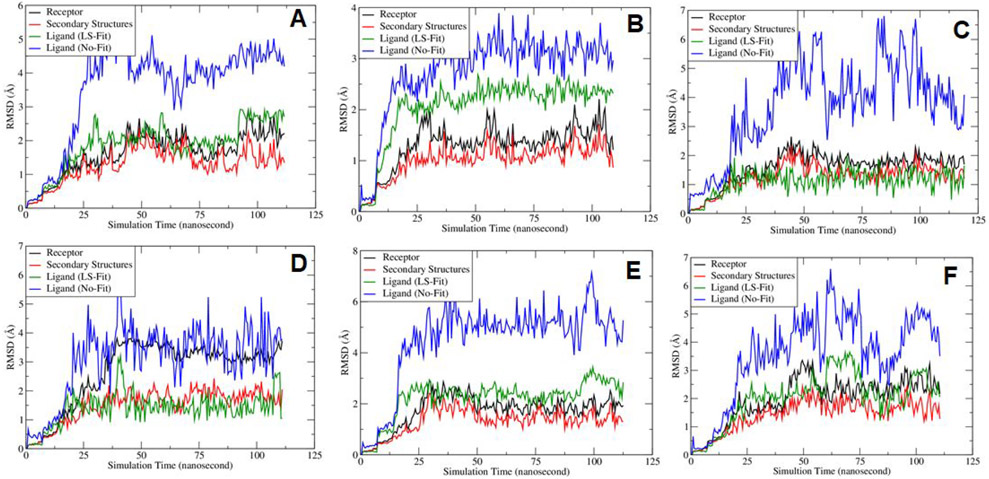
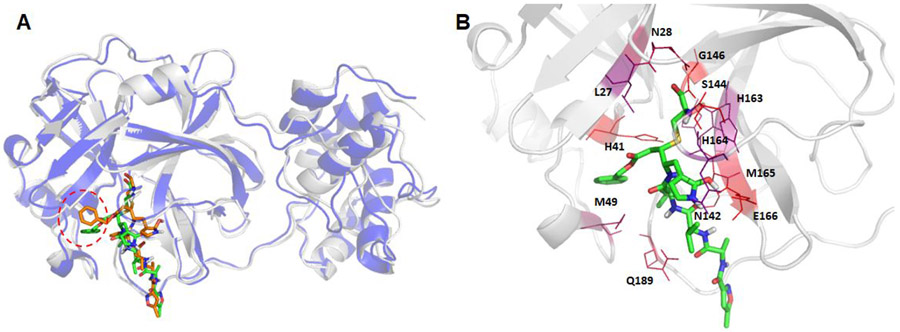
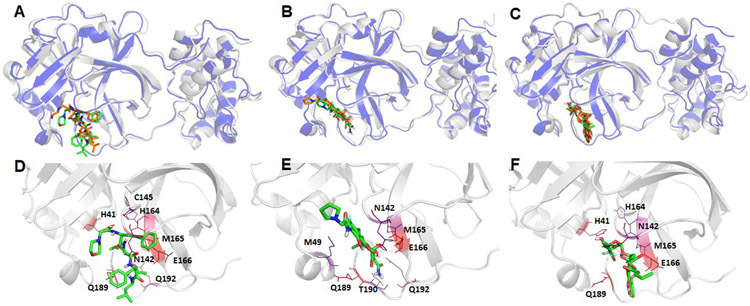
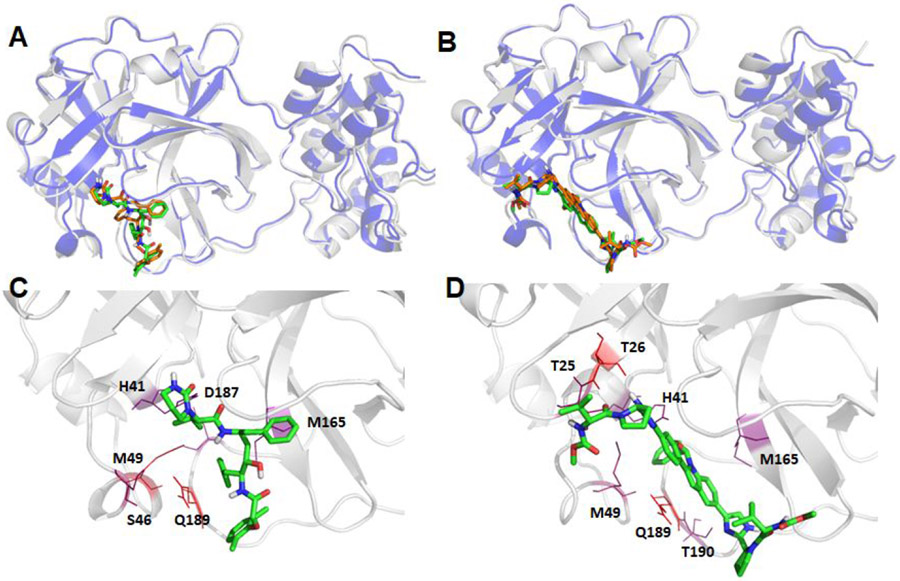
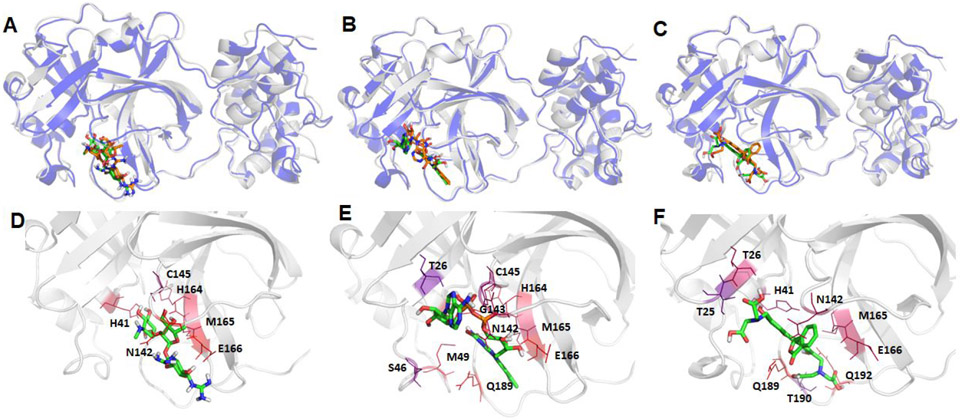
The recent outbreak of novel coronavirus disease-19 (COVID-19) calls for and welcomes possible treatment strategies using drugs on the market. It is very efficient to apply computer-aided drug design techniques to quickly identify promising drug repurposing candidates, especially after the detailed 3D structures of key viral proteins are resolved. The virus causing COVID-19 is SARS-CoV-2. Taking advantage of a recently released crystal structure of SARS-CoV-2 main protease in complex with a covalently bonded inhibitor, N3 (Liu et al., 10.2210/pdb6LU7/pdb), I conducted virtual docking screening of approved drugs and drug candidates in clinical trials. For the top docking hits, I then performed molecular dynamics simulations followed by binding free energy calculations using an end point method called MM-PBSA-WSAS (molecular mechanics/Poisson-Boltzmann surface area/weighted solvent-accessible surface area; Wang, Chem. Rev. 2019, 119, 9478; Wang, Curr. Comput.-Aided Drug Des. 2006, 2, 287; Wang; ; Hou J. Chem. Inf. Model., 2012, 52, 1199). Several promising known drugs stand out as potential inhibitors of SARS-CoV-2 main protease, including carfilzomib, eravacycline, valrubicin, lopinavir, and elbasvir. Carfilzomib, an approved anticancer drug acting as a proteasome inhibitor, has the best MM-PBSA-WSAS binding free energy, -13.8 kcal/mol. The second-best repurposing drug candidate, eravacycline, is synthetic halogenated tetracycline class antibiotic. Streptomycin, another antibiotic and a charged molecule, also demonstrates some inhibitory effect, even though the predicted binding free energy of the charged form (-3.8 kcal/mol) is not nearly as low as that of the neutral form (-7.9 kcal/mol). One bioactive, PubChem 23727975, has a binding free energy of -12.9 kcal/mol. Detailed receptor-ligand interactions were analyzed and hot spots for the receptor-ligand binding were identified. I found that one hot spot residue, His41, is a conserved residue across many viruses including SARS-CoV, SARS-CoV-2, MERS-CoV, and hepatitis C virus (HCV). The findings of this study can facilitate rational drug design targeting the SARS-CoV-2 main protease.
Figures
Update of
-
Fast Identification of Possible Drug Treatment of Coronavirus Disease -19 (COVID-19) Through Computational Drug Repurposing Study.ChemRxiv [Preprint]. 2020 Feb 21. doi: 10.26434/chemrxiv.11875446. ChemRxiv. 2020. Update in: J Chem Inf Model. 2020 Jun 22;60(6):3277-3286. doi: 10.1021/acs.jcim.0c00179. PMID: 32510523 Free PMC article. Updated. Preprint.
Comment in
-
Cancer Labs Pivot to Battle COVID-19.Cancer Discov. 2020 May;10(5):634. doi: 10.1158/2159-8290.CD-ND2020-006. Epub 2020 Mar 31. Cancer Discov. 2020. PMID: 32234716
References
-
- Liu X; Zhang B; Jin Z; Yang H; Rao Z, The crystal structure of COVID-19 main protease in complex with an inhibitor N3. 2020.
-
- Wang E; Sun H; Wang J; Wang Z; Liu H; Zhang JZH; Hou T, End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev 2019, 119, 9478–9508. - PubMed
-
- Wang JM; Hou TJ; Xu XJ, Recent Advances in Free Energy Calculations with a Combination of Molecular Mechanics and Continuum Models. Curr. Comput.-Aided Drug Des 2006, 2, 287–306.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
