

HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models
- PMID: 32315286
- PMCID: PMC7324177
- DOI: 10.1172/JCI129049
HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models
Abstract
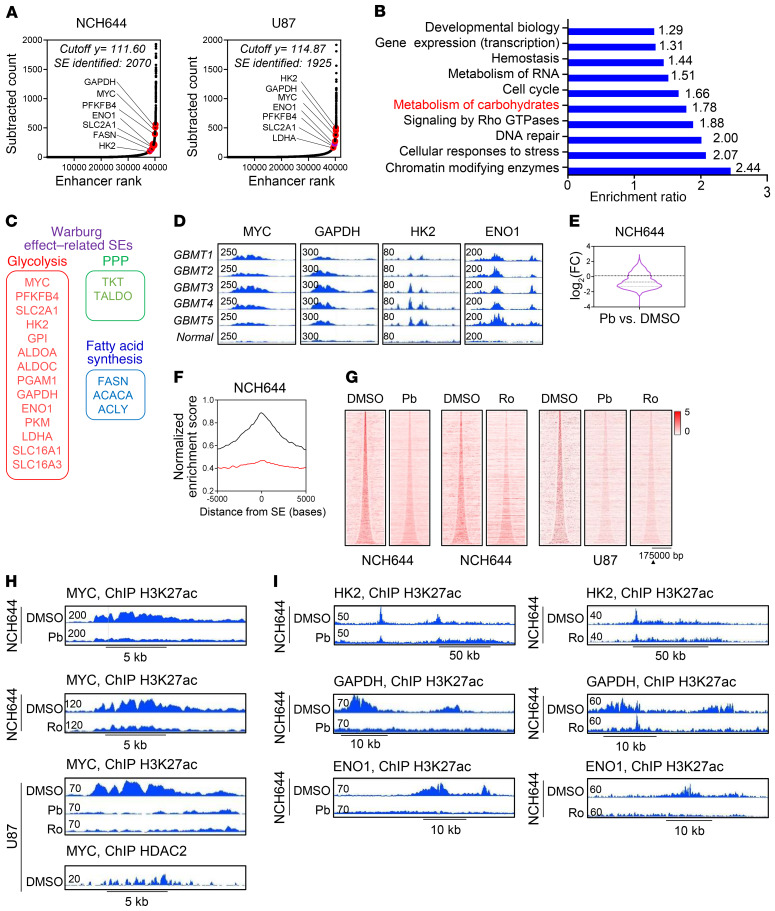
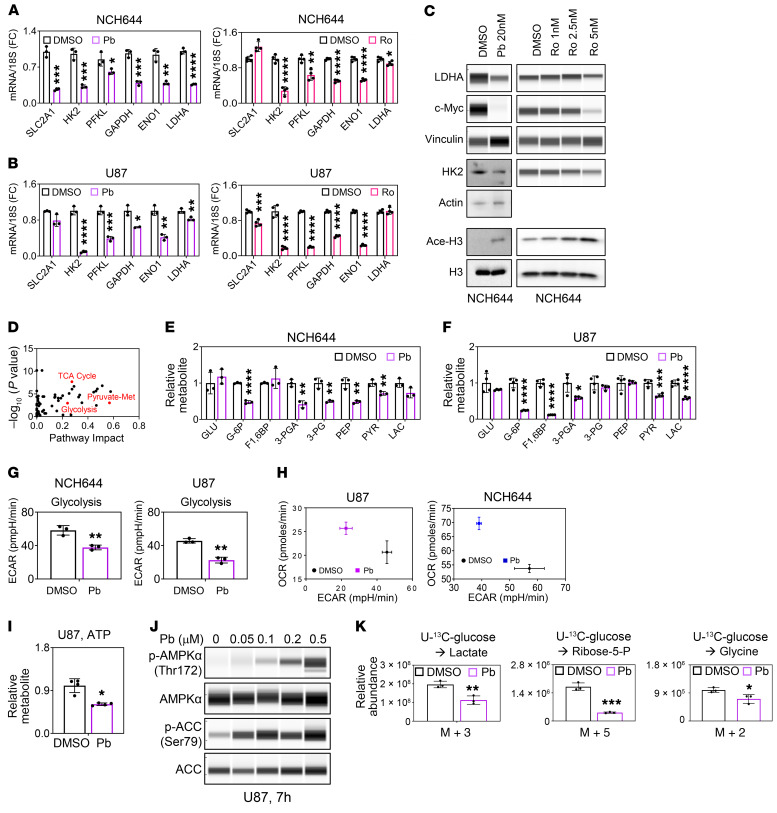
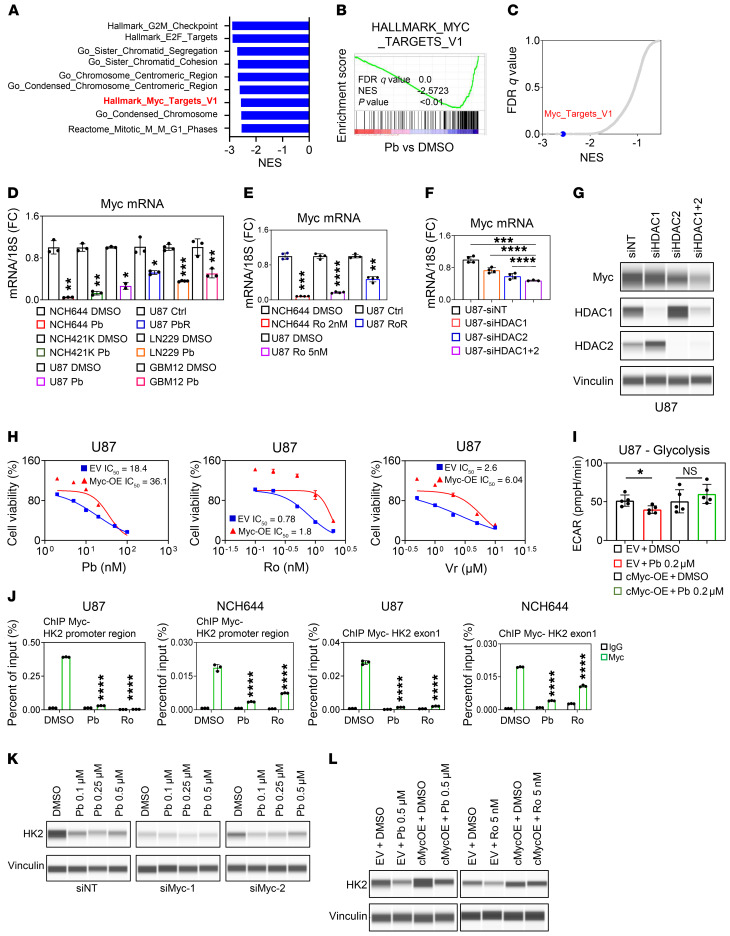
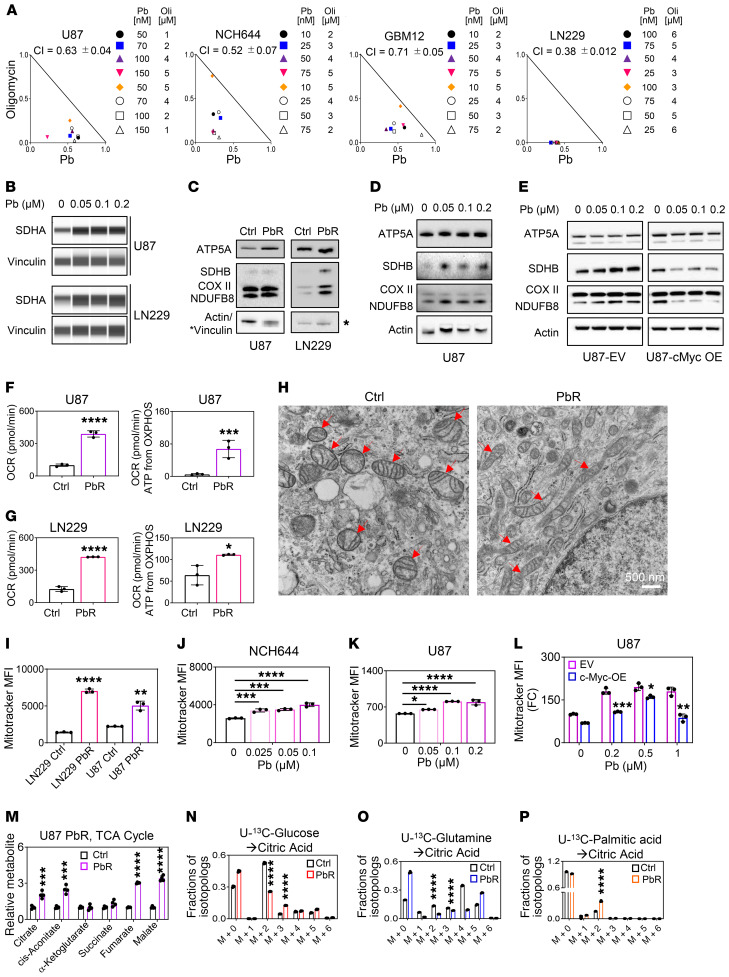
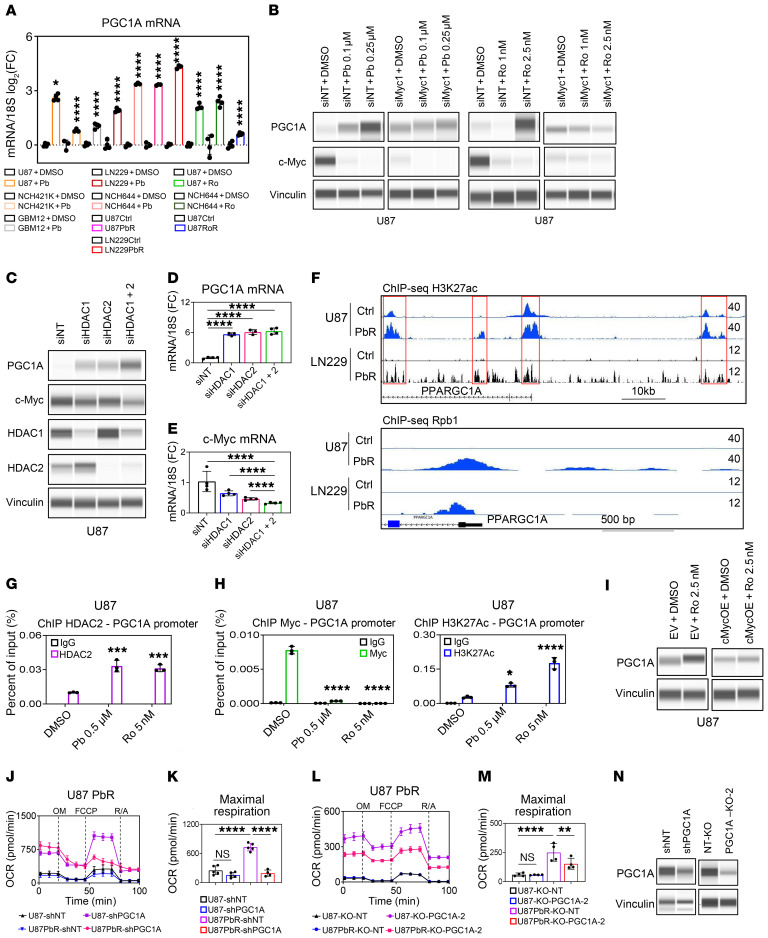
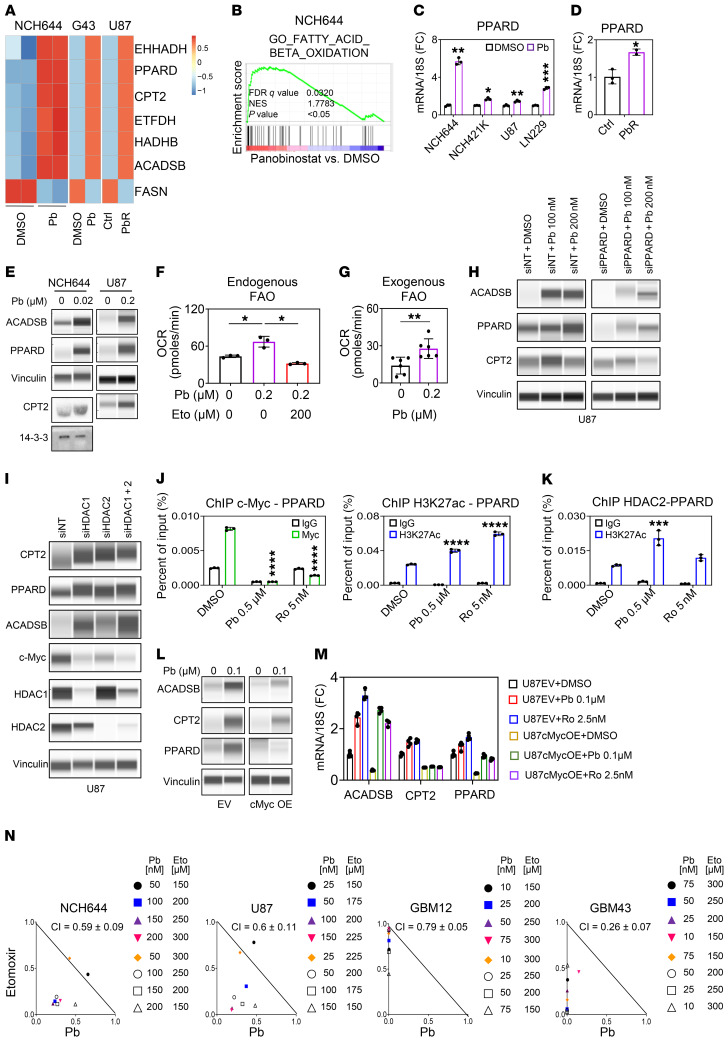
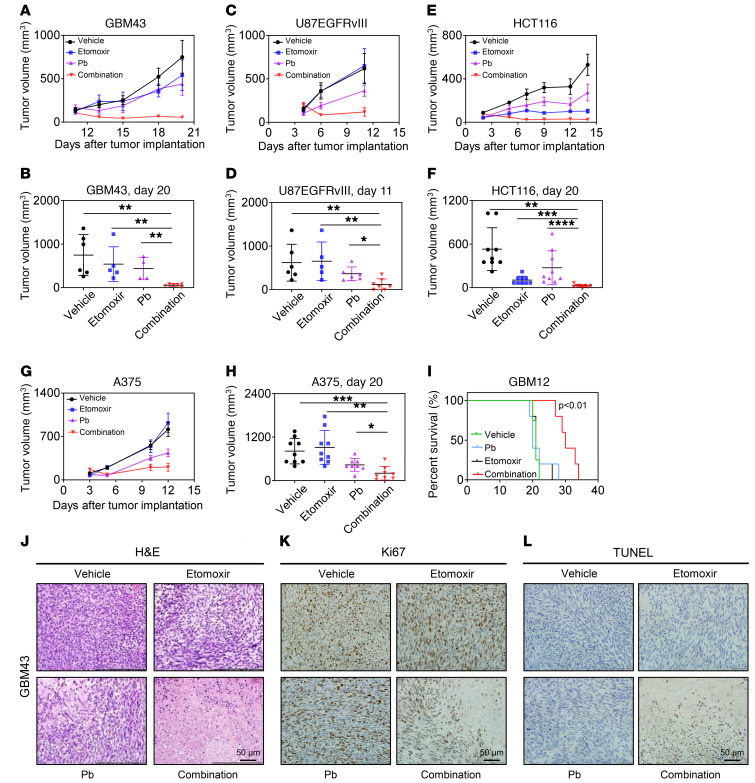
The Warburg effect is a tumor-related phenomenon that could potentially be targeted therapeutically. Here, we showed that glioblastoma (GBM) cultures and patients' tumors harbored super-enhancers in several genes related to the Warburg effect. By conducting a transcriptome analysis followed by ChIP-Seq coupled with a comprehensive metabolite analysis in GBM models, we found that FDA-approved global (panobinostat, vorinostat) and selective (romidepsin) histone deacetylase (HDAC) inhibitors elicited metabolic reprogramming in concert with disruption of several Warburg effect-related super-enhancers. Extracellular flux and carbon-tracing analyses revealed that HDAC inhibitors blunted glycolysis in a c-Myc-dependent manner and lowered ATP levels. This resulted in the engagement of oxidative phosphorylation (OXPHOS) driven by elevated fatty acid oxidation (FAO), rendering GBM cells dependent on these pathways. Mechanistically, interference with HDAC1/-2 elicited a suppression of c-Myc protein levels and a concomitant increase in 2 transcriptional drivers of oxidative metabolism, PGC1α and PPARD, suggesting an inverse relationship. Rescue and ChIP experiments indicated that c-Myc bound to the promoter regions of PGC1α and PPARD to counteract their upregulation driven by HDAC1/-2 inhibition. Finally, we demonstrated that combination treatment with HDAC and FAO inhibitors extended animal survival in patient-derived xenograft model systems in vivo more potently than single treatments in the absence of toxicity.
Keywords: Intermediary metabolism; Oncology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
