

Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity
- PMID: 32316562
- PMCID: PMC7215408
- DOI: 10.3390/ijms21082802
Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity
Abstract
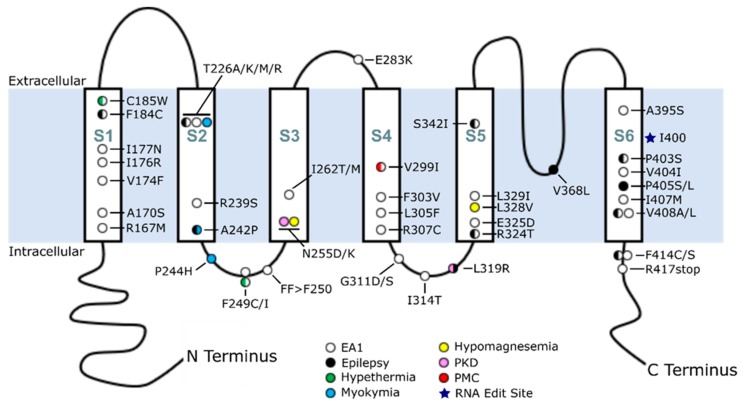
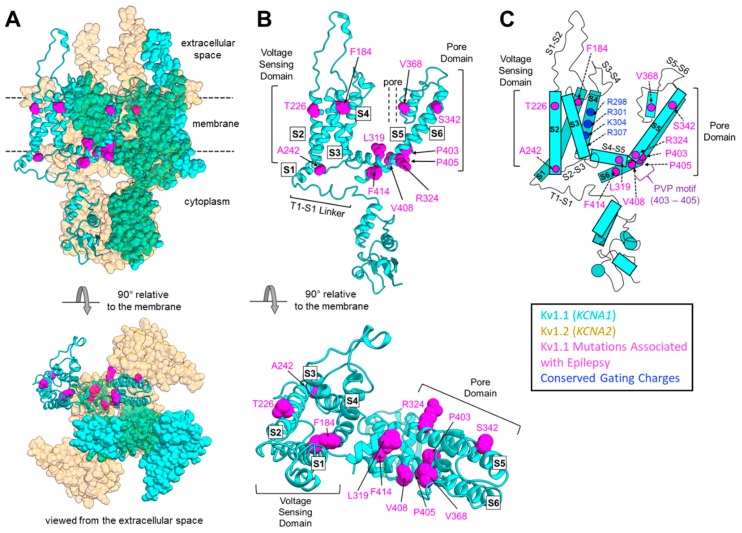
Mutations in the KCNA1 gene, which encodes voltage-gated Kv1.1 potassium channel α-subunits, cause a variety of human diseases, complicating simple genotype-phenotype correlations in patients. KCNA1 mutations are primarily associated with a rare neurological movement disorder known as episodic ataxia type 1 (EA1). However, some patients have EA1 in combination with epilepsy, whereas others have epilepsy alone. KCNA1 mutations can also cause hypomagnesemia and paroxysmal dyskinesia in rare cases. Why KCNA1 variants are associated with such phenotypic heterogeneity in patients is not yet understood. In this review, literature databases (PubMed) and public genetic archives (dbSNP and ClinVar) were mined for known pathogenic or likely pathogenic mutations in KCNA1 to examine whether patterns exist between mutation type and disease manifestation. Analyses of the 47 deleterious KCNA1 mutations that were identified revealed that epilepsy or seizure-related variants tend to cluster in the S1/S2 transmembrane domains and in the pore region of Kv1.1, whereas EA1-associated variants occur along the whole length of the protein. In addition, insights from animal models of KCNA1 channelopathy were considered, as well as the possible influence of genetic modifiers on disease expressivity and severity. Elucidation of the complex relationship between KCNA1 variants and disease will enable better diagnostic risk assessment and more personalized therapeutic strategies for KCNA1 channelopathy.
Keywords: KCNA1; Kv1.1; epilepsy; episodic ataxia.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
