Timing the initiation of multiple myeloma
- PMID: 32317634
- PMCID: PMC7174344
- DOI: 10.1038/s41467-020-15740-9
Timing the initiation of multiple myeloma
Abstract
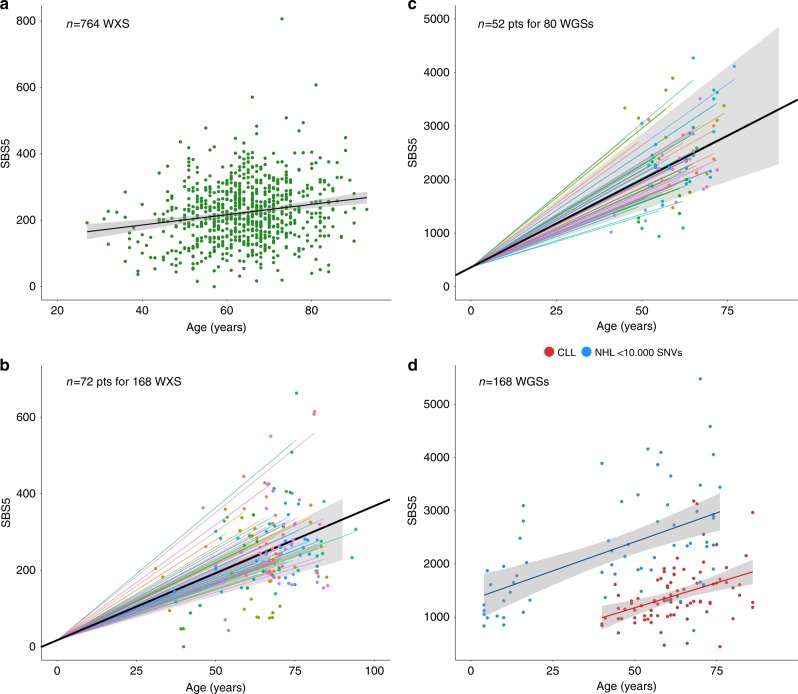
The evolution and progression of multiple myeloma and its precursors over time is poorly understood. Here, we investigate the landscape and timing of mutational processes shaping multiple myeloma evolution in a large cohort of 89 whole genomes and 973 exomes. We identify eight processes, including a mutational signature caused by exposure to melphalan. Reconstructing the chronological activity of each mutational signature, we estimate that the initial transformation of a germinal center B-cell usually occurred during the first 2nd-3rd decades of life. We define four main patterns of activation-induced deaminase (AID) and apolipoprotein B mRNA editing catalytic polypeptide-like (APOBEC) mutagenesis over time, including a subset of patients with evidence of prolonged AID activity during the pre-malignant phase, indicating antigen-responsiveness and germinal center reentry. Our findings provide a framework to study the etiology of multiple myeloma and explore strategies for prevention and early detection.
Conflict of interest statement
The authors declare no competing interests.
Figures