

Identification of novel USH2A mutations in patients with autosomal recessive retinitis pigmentosa via targeted next‑generation sequencing
- PMID: 32319668
- PMCID: PMC7248525
- DOI: 10.3892/mmr.2020.11087
Identification of novel USH2A mutations in patients with autosomal recessive retinitis pigmentosa via targeted next‑generation sequencing
Abstract
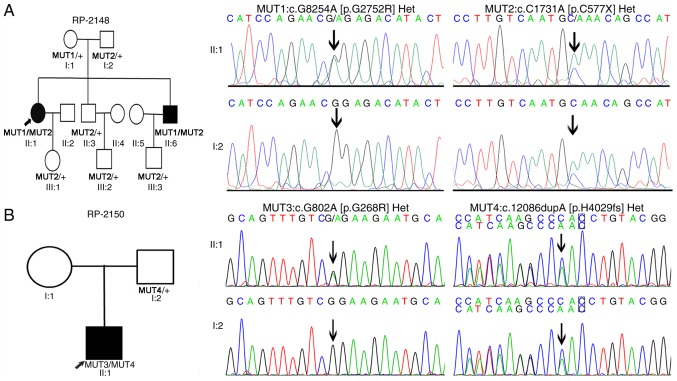
Retinitis pigmentosa (RP) is a group of inheritable blindness retinal diseases characterized by the death of photoreceptor cells and a gradual loss of peripheral vision. Mutations in Usher syndrome type 2 (USH2A) have been reported in RP with or without hearing loss. The present study aimed to identify causative mutations in a cohort of families with RP from China. A cohort of 62 non‑syndromic families with RP and 30 sporadic cases were enrolled in this study. All affected members underwent a complete ophthalmic examination, including fundus photography, visual‑field test and optical coherence tomography examination. Next‑generation sequencing‑targeted sequencing of 163 genes involved in inheritable retinal disorders was performed on the probands. Stringent bioinformatics data analysis was applied to identify potential candidate variants. In total, 6 novel mutations and 2 known mutations of USH2A were identified in 4 families with RP. A stop‑gain mutation (c.C1731A) and a missense mutation (c.G8254A) were identified in RP family RP‑2148. In another RP family, RP‑2150, a known mutation (c.G802A) and a novel frameshift insertion mutation (c.12086dupA) were discovered. A novel stop‑gain mutation (c.G11754A) and a missense mutation (c.G13465A) were identified in family rpz05. A novel missense mutation (c.C9328G) and a known missense mutation (c.G8232C) were also identified. These mutations were subsequently confirmed by Sanger sequencing. All 6 novel mutations affected highly conserved amino acid residues, and were absent in 1,000 ethnically matched controls. Taken together, the present study has reported on 6 novel USH2A mutations in 4 families with RP, and has expanded the mutation spectrum of USH2A in autosomal recessive RP in the Chinese population, thus providing important information for the molecular diagnosis and screening of RP.
Keywords: targeted sequencing; ene mutation; usher syndrome type 2; USH2A; retinitis pigmentosa.
Figures




References
-
- Dryja TP, Hahn LB, Kajiwara K, Berson EL. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1997;38:1972–1982. - PubMed
