

Defective Influenza A Virus RNA Products Mediate MAVS-Dependent Upregulation of Human Leukocyte Antigen Class I Proteins
- PMID: 32321802
- PMCID: PMC7307169
- DOI: 10.1128/JVI.00165-20
Defective Influenza A Virus RNA Products Mediate MAVS-Dependent Upregulation of Human Leukocyte Antigen Class I Proteins
Abstract
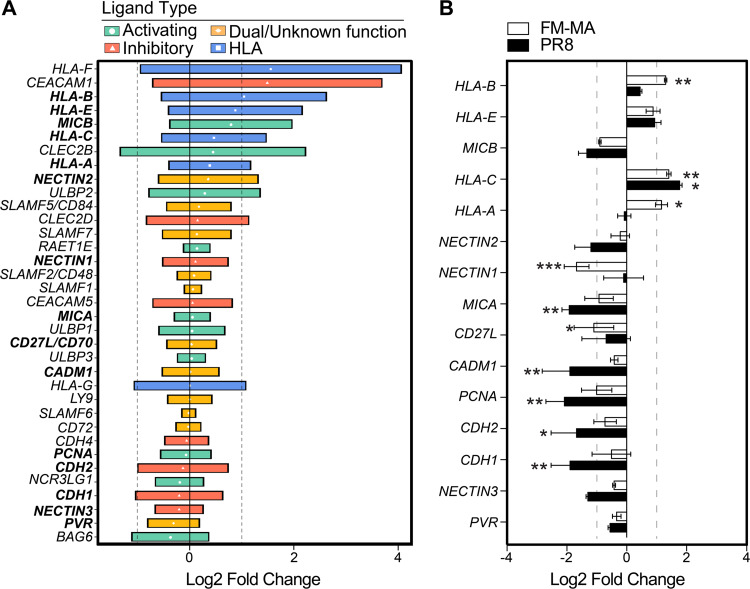
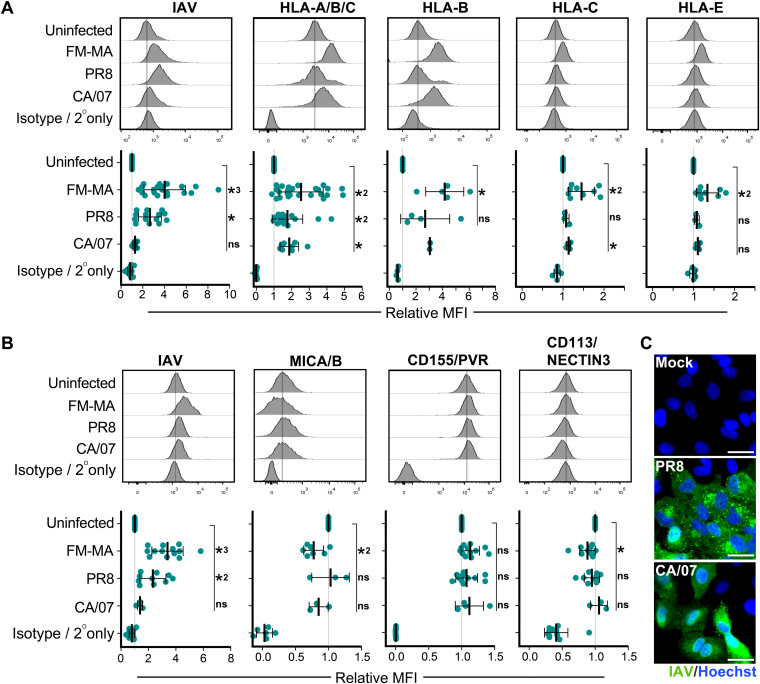
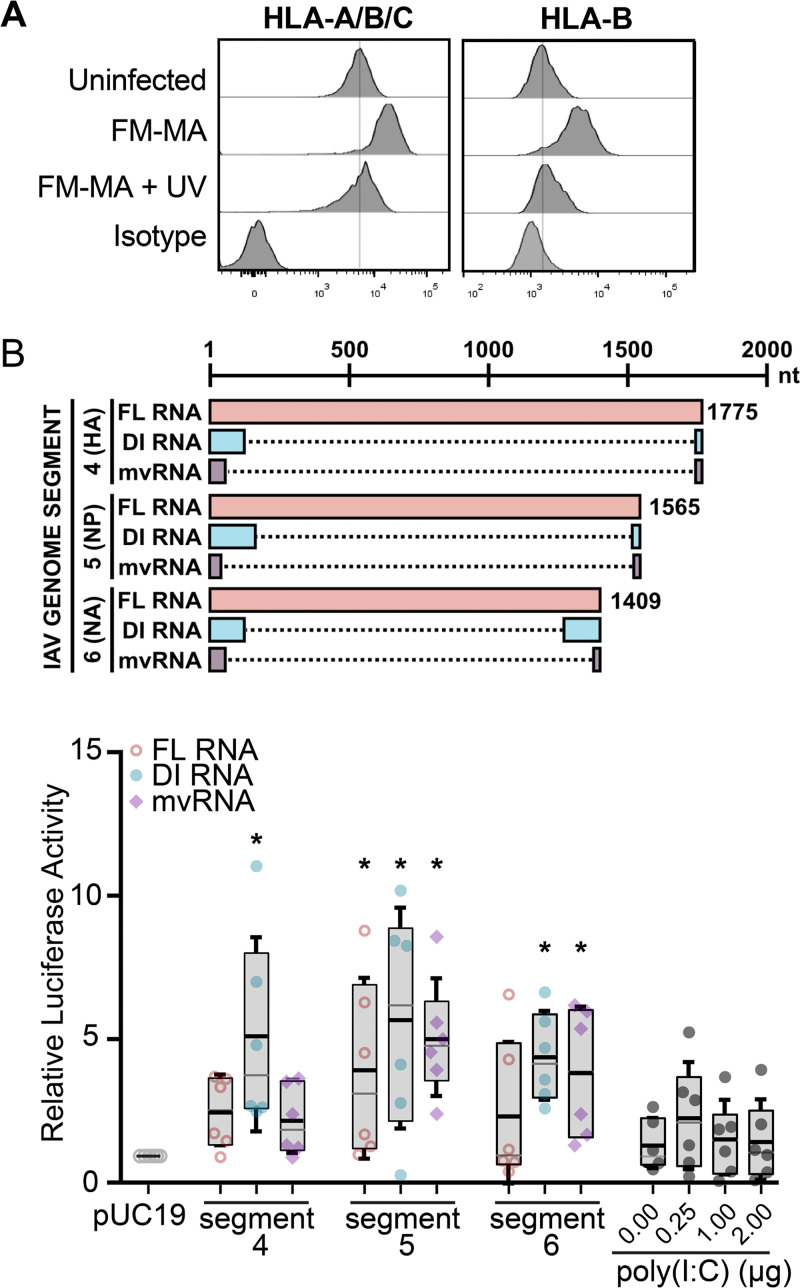
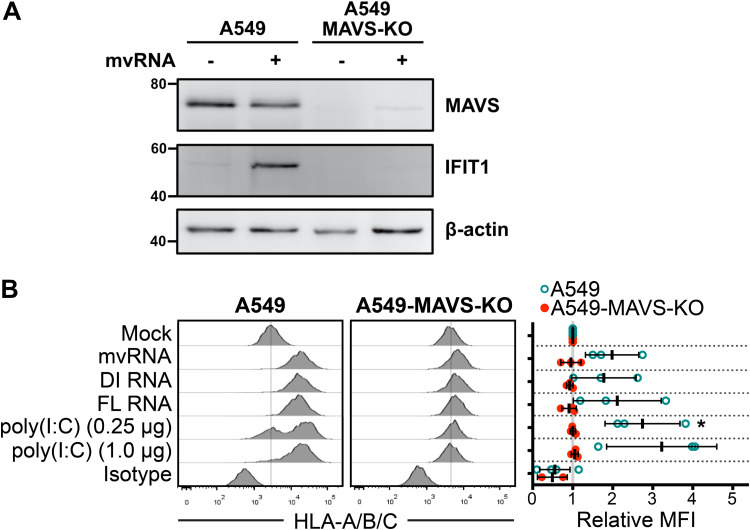
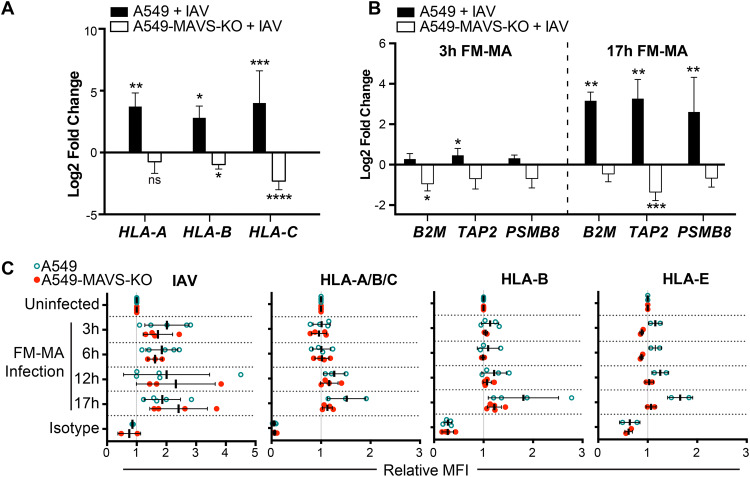
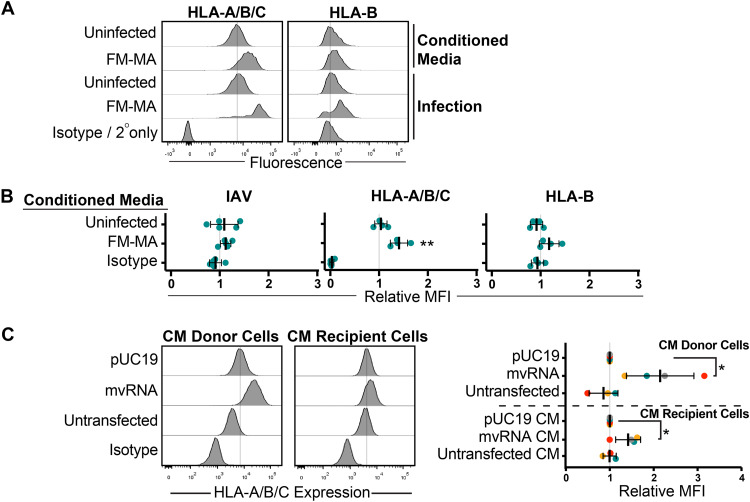
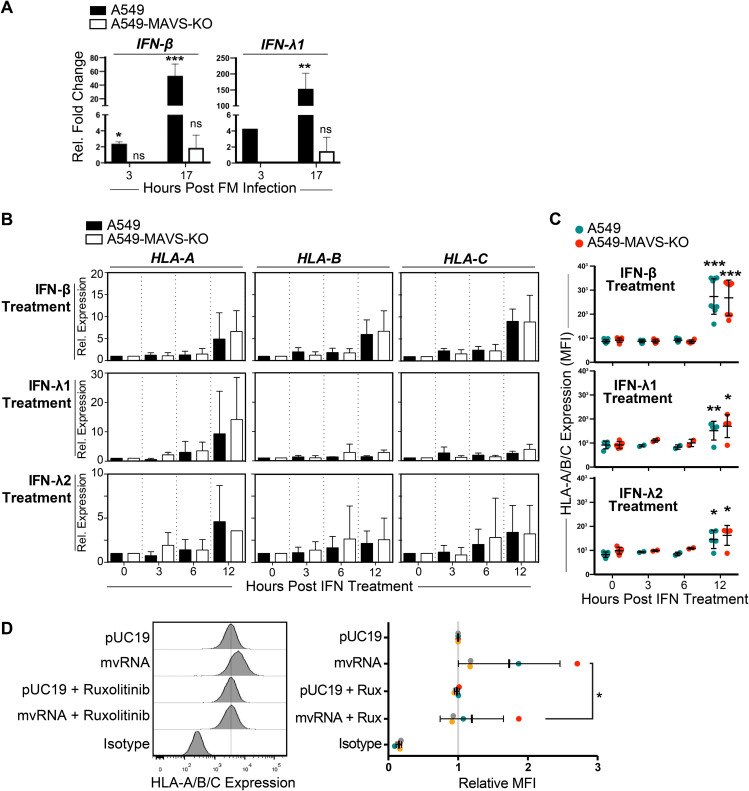
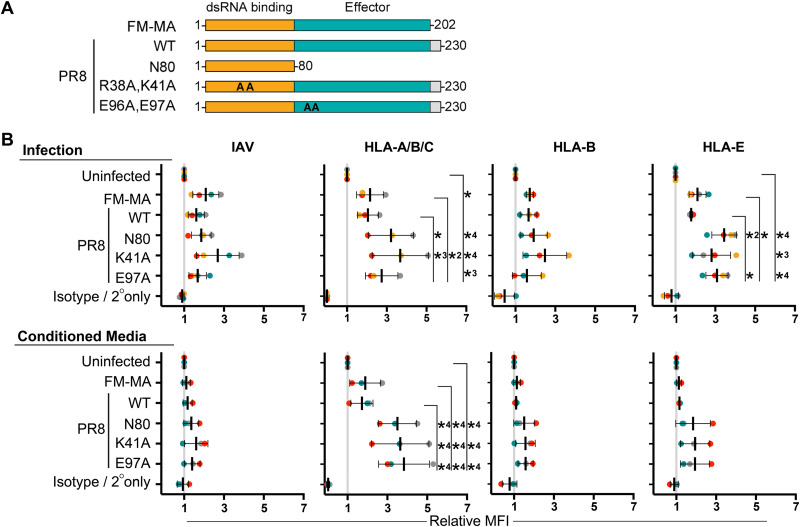
Influenza A virus (IAV) increases the presentation of class I human leukocyte antigen (HLA) proteins that limit antiviral responses mediated by natural killer (NK) cells, but molecular mechanisms for these processes have not yet been fully elucidated. We observed that infection with A/Fort Monmouth/1/1947(H1N1) IAV significantly increased the presentation of HLA-B, -C, and -E on lung epithelial cells. Virus entry was not sufficient to induce HLA upregulation because UV-inactivated virus had no effect. Aberrant internally deleted viral RNAs (vRNAs) known as mini viral RNAs (mvRNAs) and defective interfering RNAs (DI RNAs) expressed from an IAV minireplicon were sufficient for inducing HLA upregulation. These defective RNAs bind to retinoic acid-inducible gene I (RIG-I) and initiate mitochondrial antiviral signaling (MAVS) protein-dependent antiviral interferon (IFN) responses. Indeed, MAVS was required for HLA upregulation in response to IAV infection or ectopic mvRNA/DI RNA expression. The effect was partially due to paracrine signaling, as we observed that IAV infection or mvRNA/DI RNA-expression stimulated production of IFN-β and IFN-λ1 and conditioned media from these cells elicited a modest increase in HLA surface levels in naive epithelial cells. HLA upregulation in response to aberrant viral RNAs could be prevented by the Janus kinase (JAK) inhibitor ruxolitinib. While HLA upregulation would seem to be advantageous to the virus, it is kept in check by the viral nonstructural 1 (NS1) protein; we determined that NS1 limits cell-intrinsic and paracrine mechanisms of HLA upregulation. Taken together, our findings indicate that aberrant IAV RNAs stimulate HLA presentation, which may aid viral evasion of innate immunity.IMPORTANCE Human leukocyte antigens (HLAs) are cell surface proteins that regulate innate and adaptive immune responses to viral infection by engaging with receptors on immune cells. Many viruses have evolved ways to evade host immune responses by modulating HLA expression and/or processing. Here, we provide evidence that aberrant RNA products of influenza virus genome replication can trigger retinoic acid-inducible gene I (RIG-I)/mitochondrial antiviral signaling (MAVS)-dependent remodeling of the cell surface, increasing surface presentation of HLA proteins known to inhibit the activation of an immune cell known as a natural killer (NK) cell. While this HLA upregulation would seem to be advantageous to the virus, it is kept in check by the viral nonstructural 1 (NS1) protein, which limits RIG-I activation and interferon production by the infected cell.
Keywords: DI RNAs; KIR; MAVS; NK cells; RIG-I; class I HLA; class I MHC; influenza A virus; interferon; mvRNAs.
Copyright © 2020 Rahim et al.
Figures
Similar articles
-
Inhibition of Ongoing Influenza A Virus Replication Reveals Different Mechanisms of RIG-I Activation.J Virol. 2019 Mar 5;93(6):e02066-18. doi: 10.1128/JVI.02066-18. Print 2019 Mar 15. J Virol. 2019. PMID: 30602605 Free PMC article.
-
Differential Modulation of Innate Immune Responses in Human Primary Cells by Influenza A Viruses Carrying Human or Avian Nonstructural Protein 1.J Virol. 2019 Dec 12;94(1):e00999-19. doi: 10.1128/JVI.00999-19. Print 2019 Dec 12. J Virol. 2019. PMID: 31597767 Free PMC article.
-
Role of the Chaperone Protein 14-3-3ε in the Regulation of Influenza A Virus-Activated Beta Interferon.J Virol. 2021 Sep 27;95(20):e0023121. doi: 10.1128/JVI.00231-21. Epub 2021 Aug 11. J Virol. 2021. PMID: 34379499 Free PMC article.
-
Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins.Viruses. 2018 Dec 12;10(12):708. doi: 10.3390/v10120708. Viruses. 2018. PMID: 30545063 Free PMC article. Review.
-
Interplay between influenza A virus and the innate immune signaling.Microbes Infect. 2010 Jan;12(1):81-7. doi: 10.1016/j.micinf.2009.09.007. Epub 2009 Sep 25. Microbes Infect. 2010. PMID: 19782761 Review.
Cited by
-
Pathogen-driven CRISPR screens identify TREX1 as a regulator of DNA self-sensing during influenza virus infection.Cell Host Microbe. 2023 Sep 13;31(9):1552-1567.e8. doi: 10.1016/j.chom.2023.08.001. Epub 2023 Aug 30. Cell Host Microbe. 2023. PMID: 37652009 Free PMC article.
-
Transient RNA structures cause aberrant influenza virus replication and innate immune activation.Sci Adv. 2022 Sep 9;8(36):eabp8655. doi: 10.1126/sciadv.abp8655. Epub 2022 Sep 9. Sci Adv. 2022. PMID: 36083899 Free PMC article.
-
Influenza A virus NS1 effector domain is required for PA-X-mediated host shutoff in infected cells.J Virol. 2024 May 14;98(5):e0190123. doi: 10.1128/jvi.01901-23. Epub 2024 Apr 17. J Virol. 2024. PMID: 38629840 Free PMC article.
-
Positive Regulation of Cellular Proteins by Influenza Virus for Productive Infection.Int J Mol Sci. 2025 Apr 10;26(8):3584. doi: 10.3390/ijms26083584. Int J Mol Sci. 2025. PMID: 40332127 Free PMC article. Review.
-
Cytosolic nucleic acid sensing as driver of critical illness: mechanisms and advances in therapy.Signal Transduct Target Ther. 2025 Mar 19;10(1):90. doi: 10.1038/s41392-025-02174-2. Signal Transduct Target Ther. 2025. PMID: 40102400 Free PMC article. Review.
References
-
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh C-S, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi:10.1038/nature04734. - DOI - PubMed
-
- Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M. 2007. Cutting Edge: influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol 178:3368–3372. doi:10.4049/jimmunol.178.6.3368. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
