

Consistent RNA sequencing contamination in GTEx and other data sets
- PMID: 32321923
- PMCID: PMC7176728
- DOI: 10.1038/s41467-020-15821-9
Consistent RNA sequencing contamination in GTEx and other data sets
Abstract
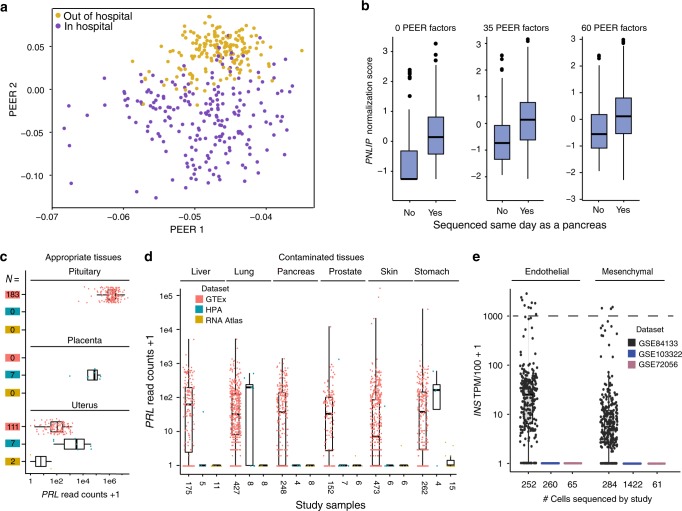
A challenge of next generation sequencing is read contamination. We use Genotype-Tissue Expression (GTEx) datasets and technical metadata along with RNA-seq datasets from other studies to understand factors that contribute to contamination. Here we report, of 48 analyzed tissues in GTEx, 26 have variant co-expression clusters of four highly expressed and pancreas-enriched genes (PRSS1, PNLIP, CLPS, and/or CELA3A). Fourteen additional highly expressed genes from other tissues also indicate contamination. Sample contamination is strongly associated with a sample being sequenced on the same day as a tissue that natively expresses those genes. Discrepant SNPs across four contaminating genes validate the contamination. Low-level contamination affects ~40% of samples and leads to numerous eQTL assignments in inappropriate tissues among these 18 genes. This type of contamination occurs widely, impacting bulk and single cell (scRNA-seq) data set analysis. In conclusion, highly expressed, tissue-enriched genes basally contaminate GTEx and other datasets impacting analyses.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
