

Mechanisms and therapeutic implications of hypermutation in gliomas
- PMID: 32322066
- PMCID: PMC8235024
- DOI: 10.1038/s41586-020-2209-9
Mechanisms and therapeutic implications of hypermutation in gliomas
Abstract
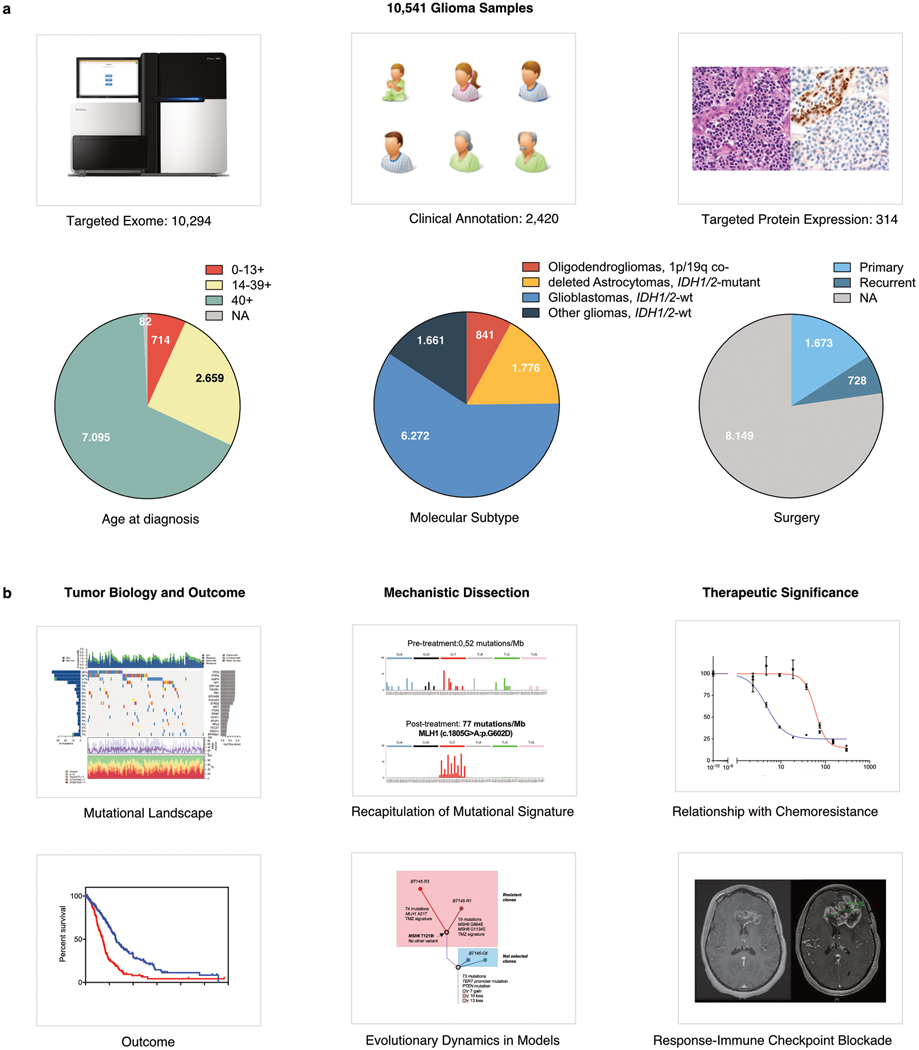
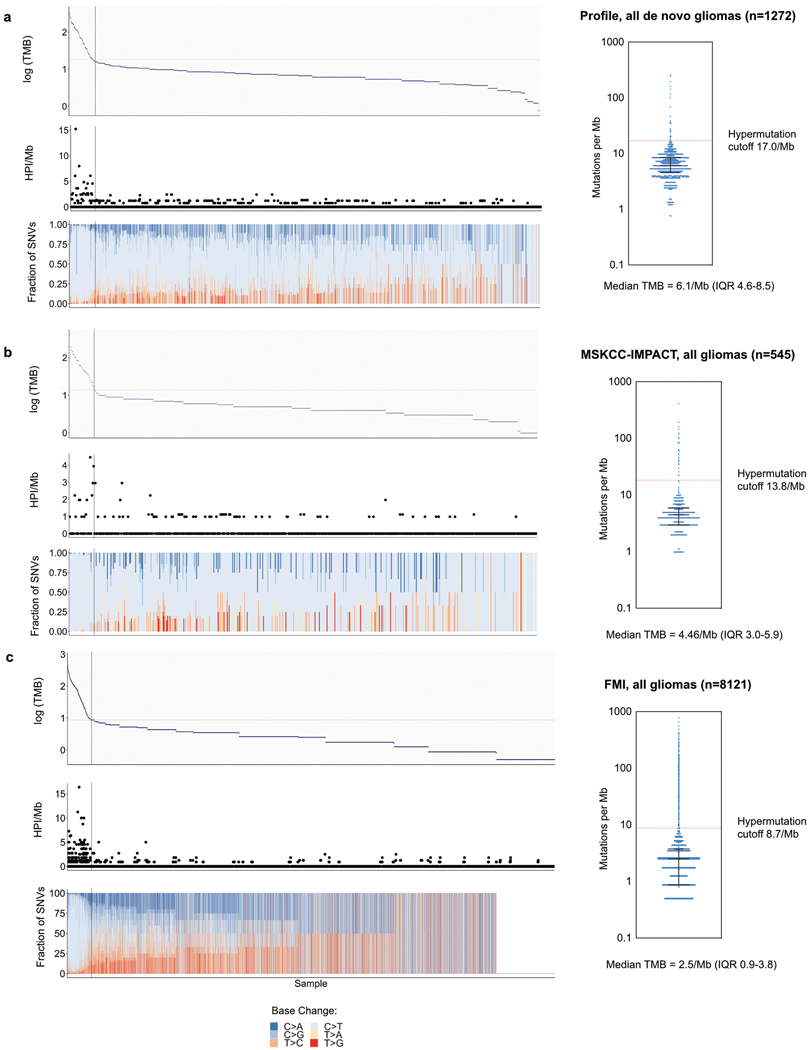
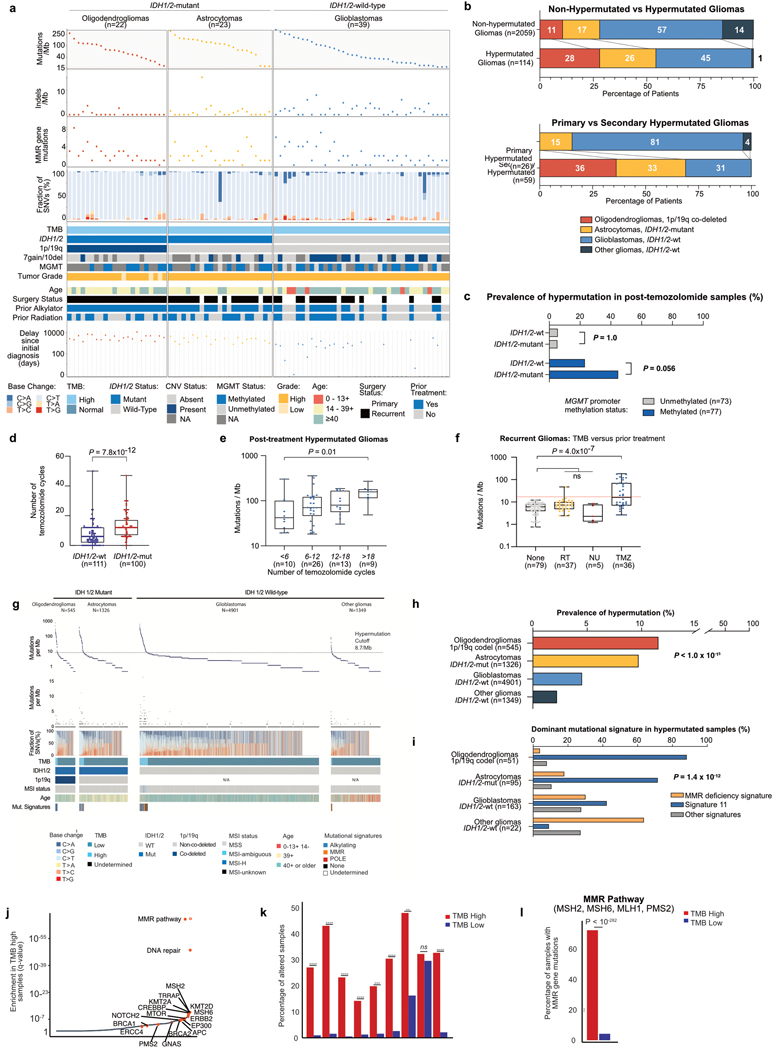
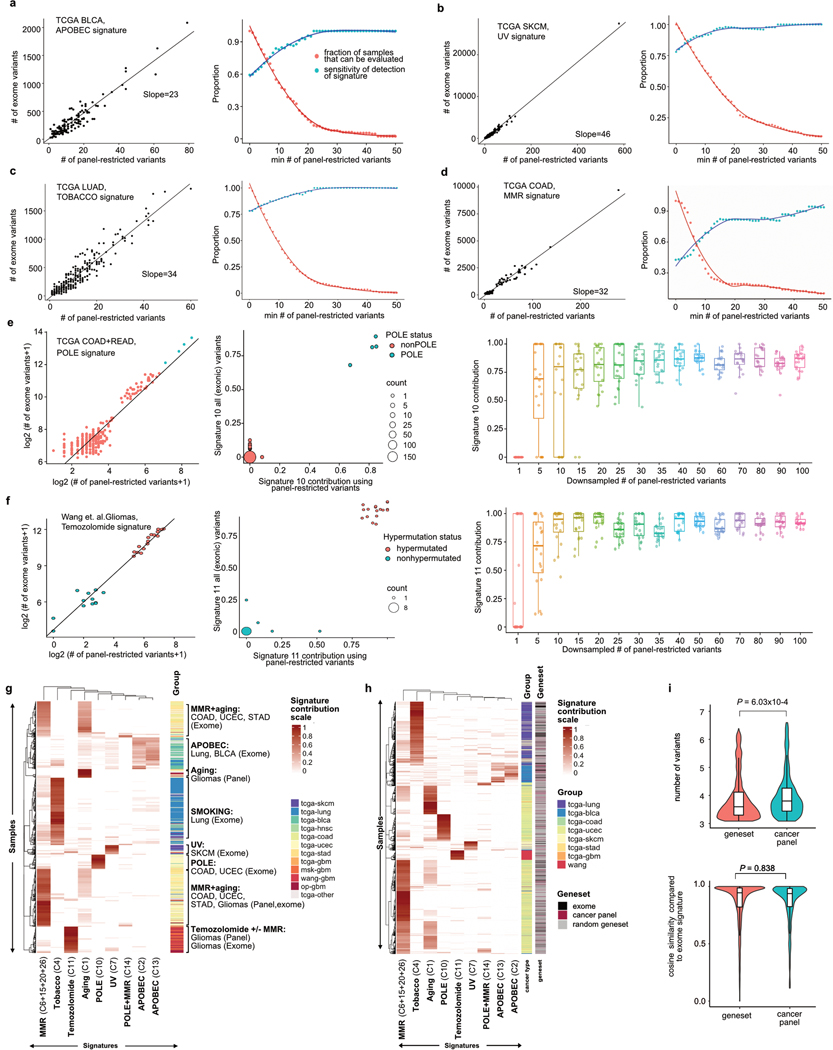
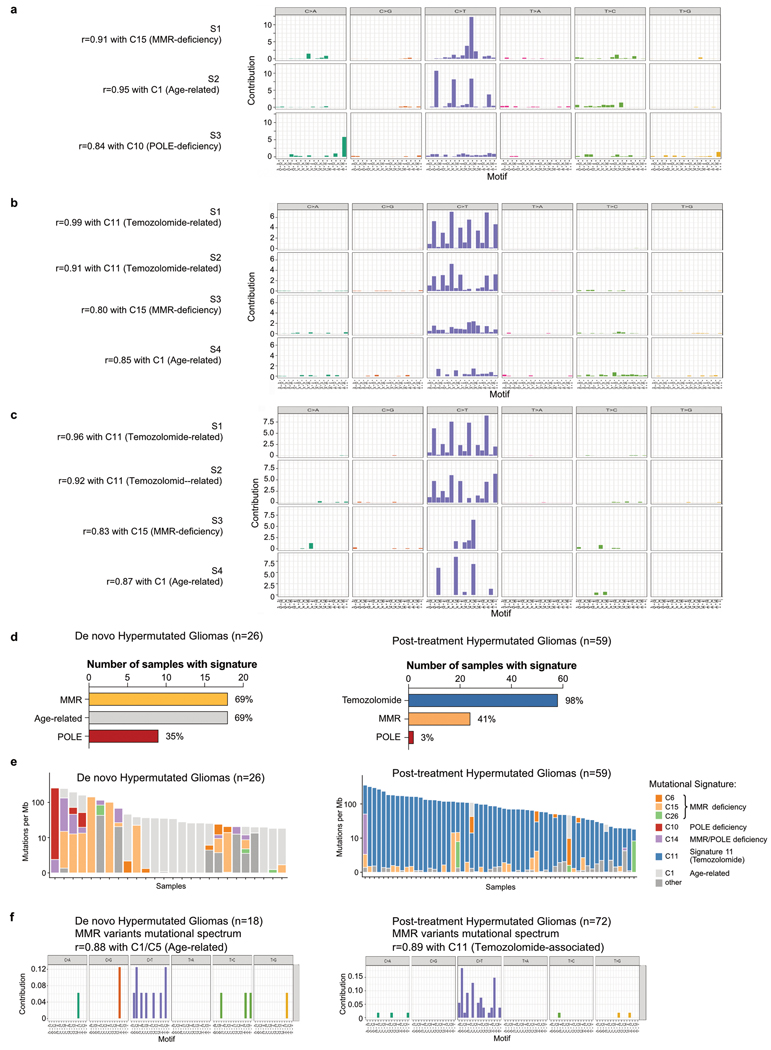
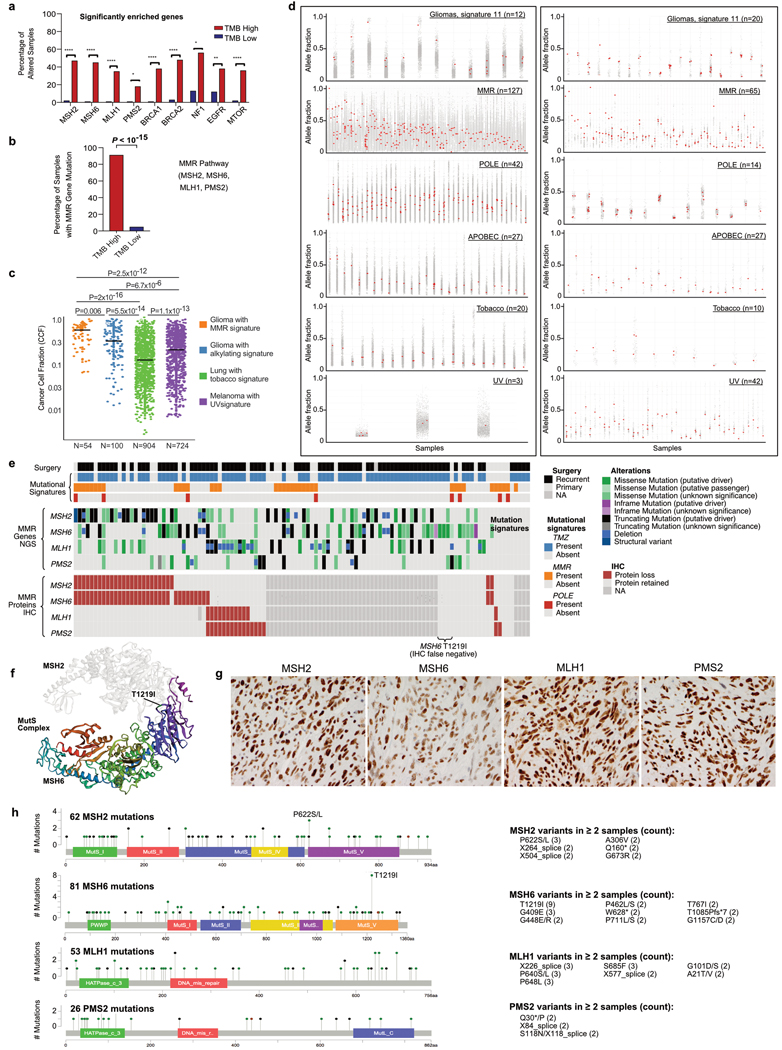
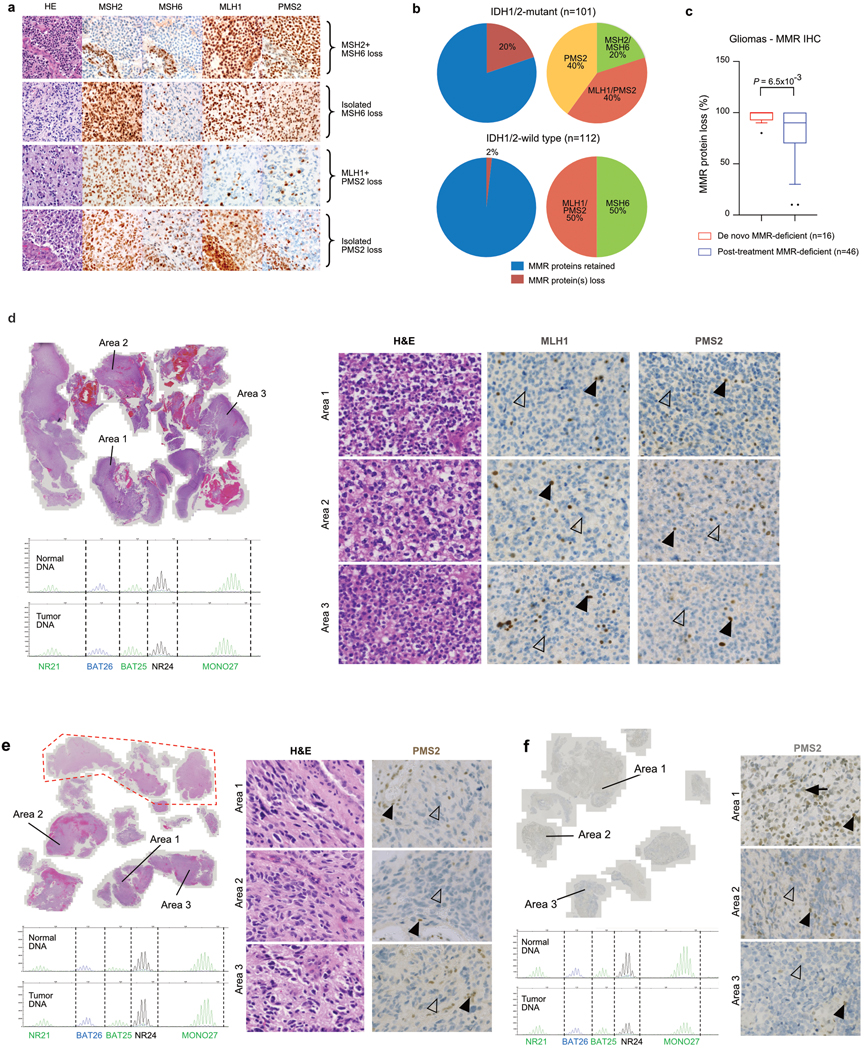
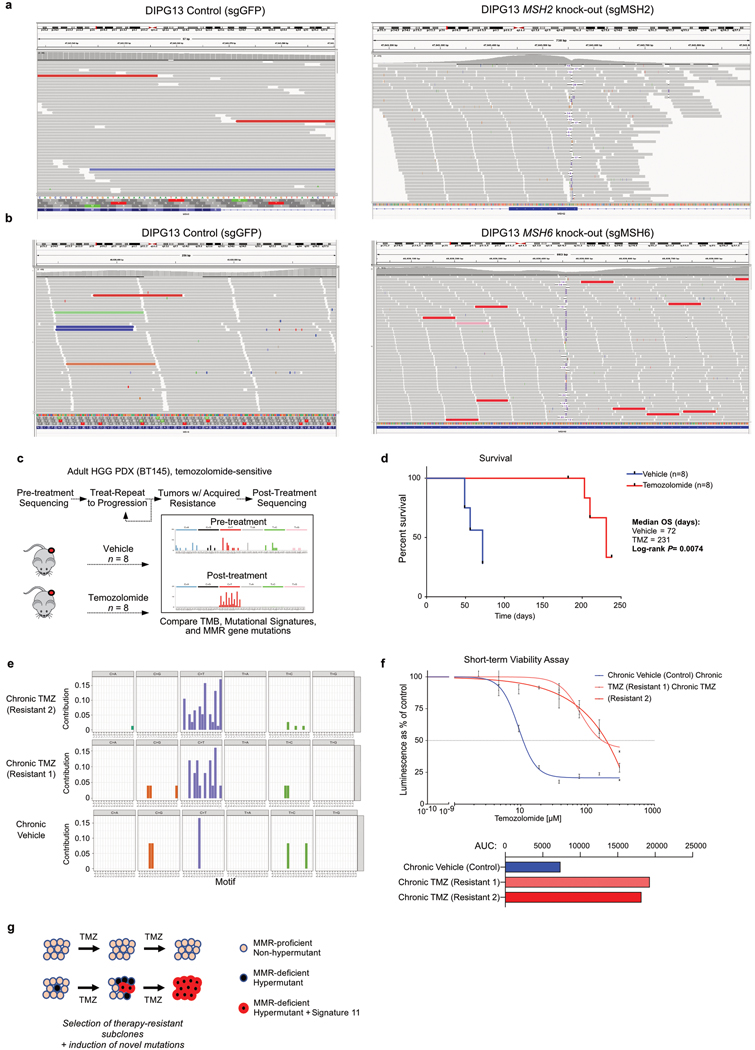
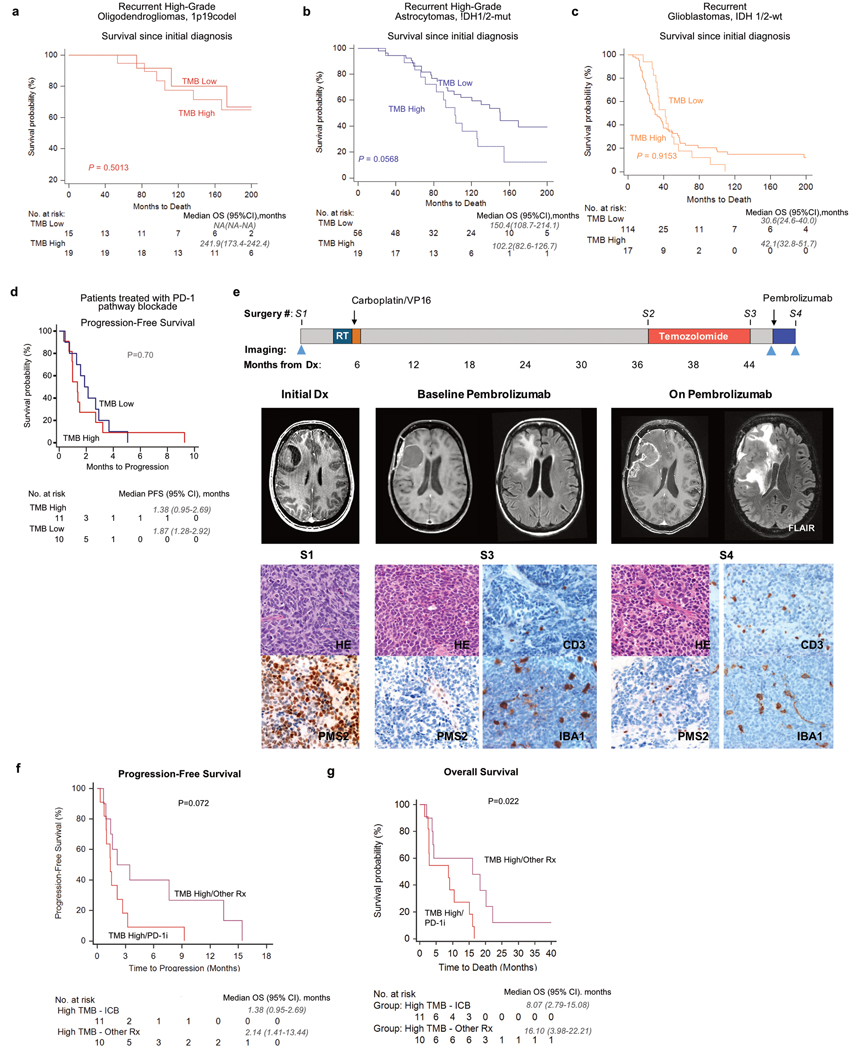
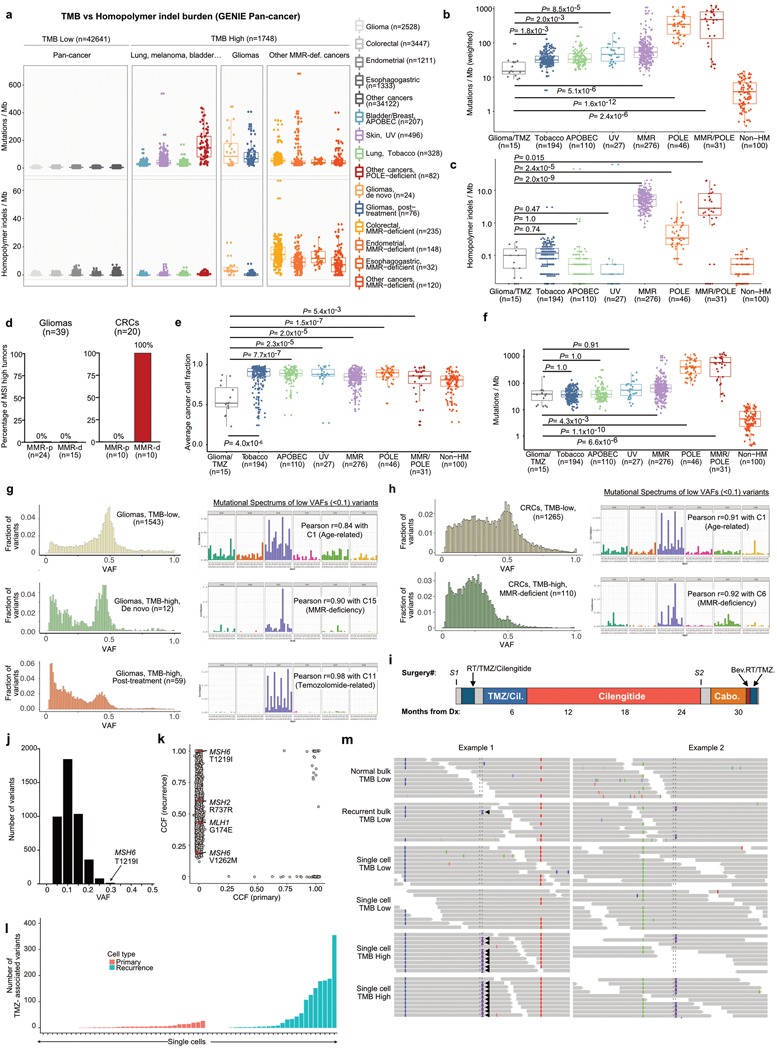
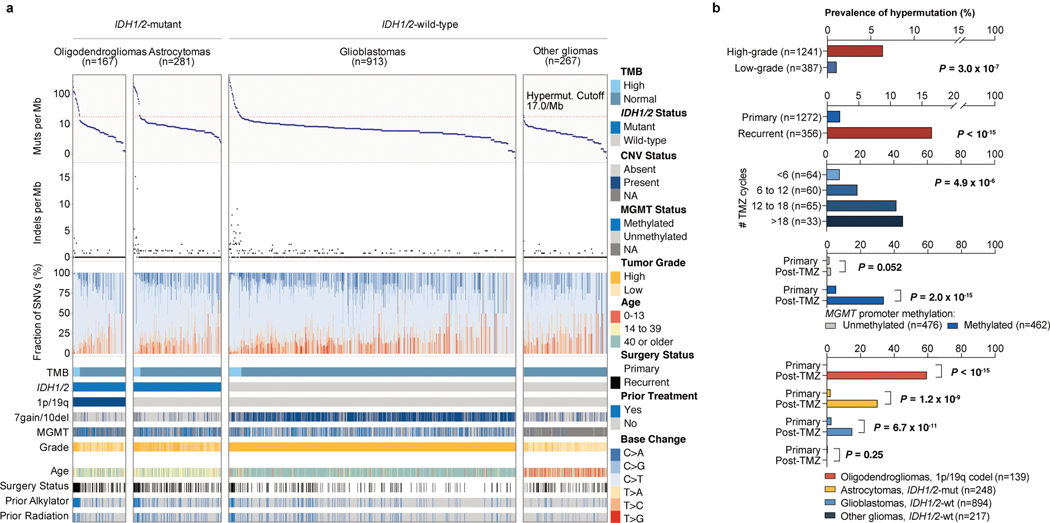
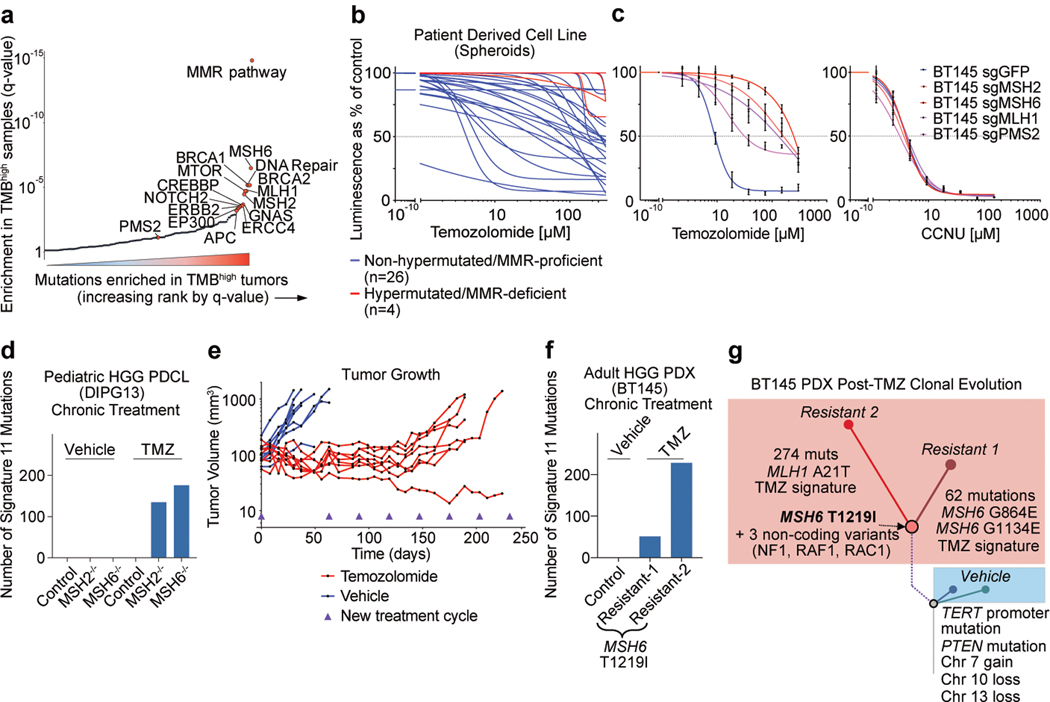
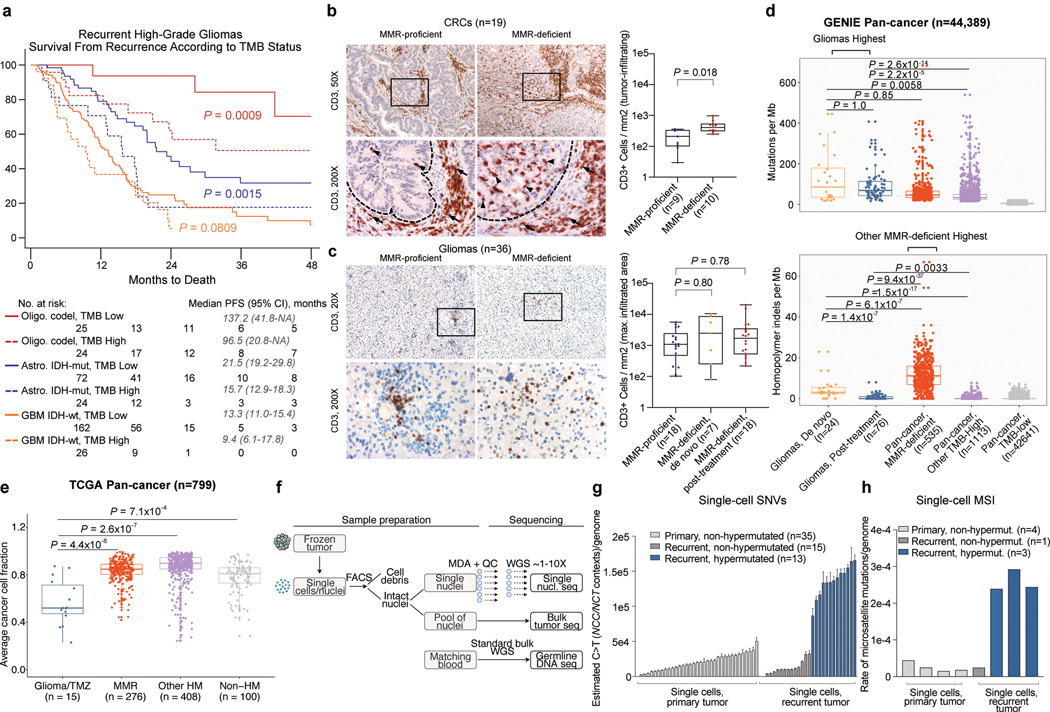
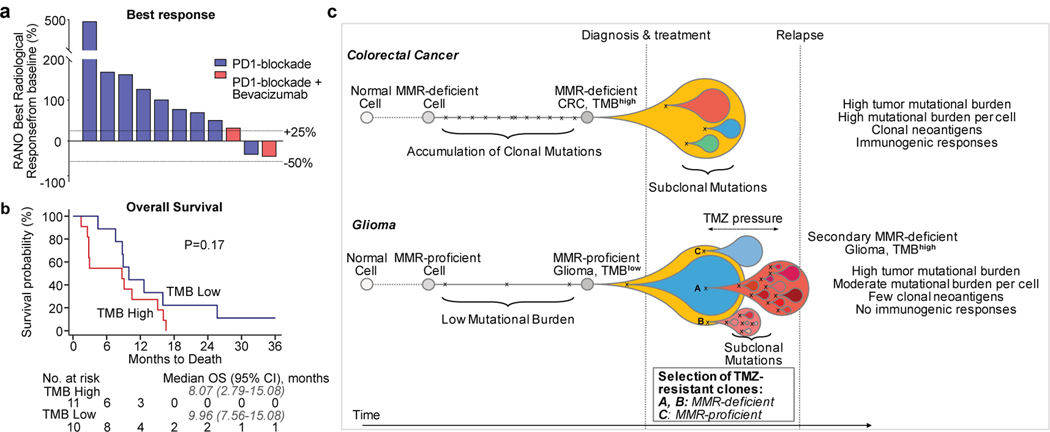
A high tumour mutational burden (hypermutation) is observed in some gliomas1-5; however, the mechanisms by which hypermutation develops and whether it predicts the response to immunotherapy are poorly understood. Here we comprehensively analyse the molecular determinants of mutational burden and signatures in 10,294 gliomas. We delineate two main pathways to hypermutation: a de novo pathway associated with constitutional defects in DNA polymerase and mismatch repair (MMR) genes, and a more common post-treatment pathway, associated with acquired resistance driven by MMR defects in chemotherapy-sensitive gliomas that recur after treatment with the chemotherapy drug temozolomide. Experimentally, the mutational signature of post-treatment hypermutated gliomas was recapitulated by temozolomide-induced damage in cells with MMR deficiency. MMR-deficient gliomas were characterized by a lack of prominent T cell infiltrates, extensive intratumoral heterogeneity, poor patient survival and a low rate of response to PD-1 blockade. Moreover, although bulk analyses did not detect microsatellite instability in MMR-deficient gliomas, single-cell whole-genome sequencing analysis of post-treatment hypermutated glioma cells identified microsatellite mutations. These results show that chemotherapy can drive the acquisition of hypermutated populations without promoting a response to PD-1 blockade and supports the diagnostic use of mutational burden and signatures in cancer.
Conflict of interest statement
The other authors report no competing interests.
Figures

Comment in
-
Hypermutated Gliomas Respond Poorly to Immunotherapy.Cancer Discov. 2020 Jul;10(7):OF5. doi: 10.1158/2159-8290.CD-NB2020-045. Epub 2020 May 20. Cancer Discov. 2020. PMID: 32434948
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
