

Non-parametric and semi-parametric support estimation using SEquential RESampling random walks on biomolecular sequences
- PMID: 32322294
- PMCID: PMC7164268
- DOI: 10.1186/s13015-020-00167-0
Non-parametric and semi-parametric support estimation using SEquential RESampling random walks on biomolecular sequences
Abstract
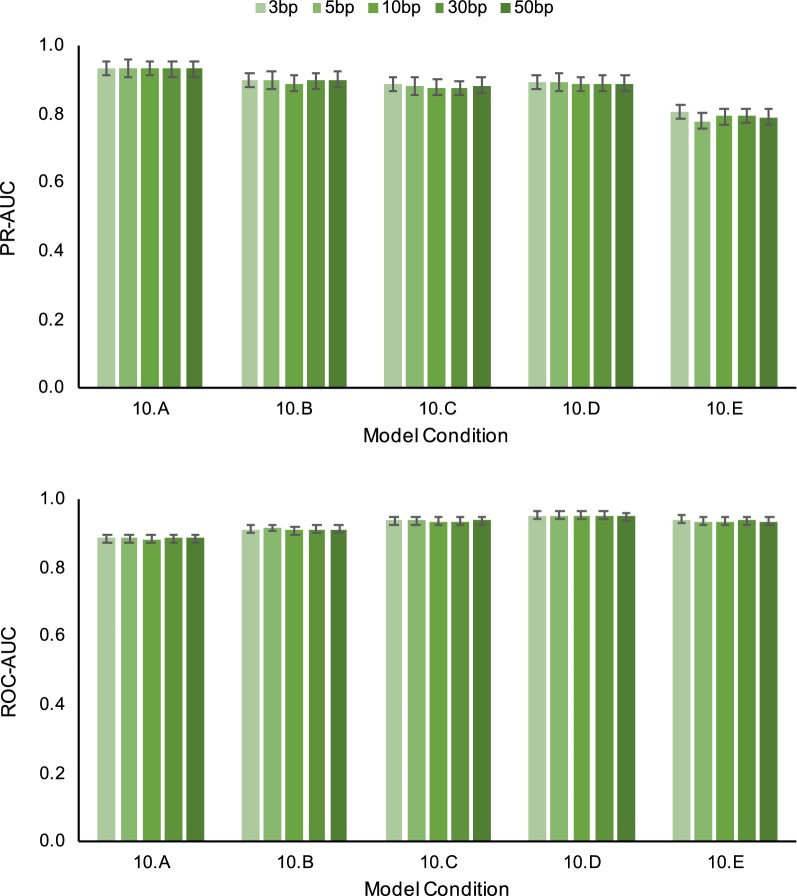
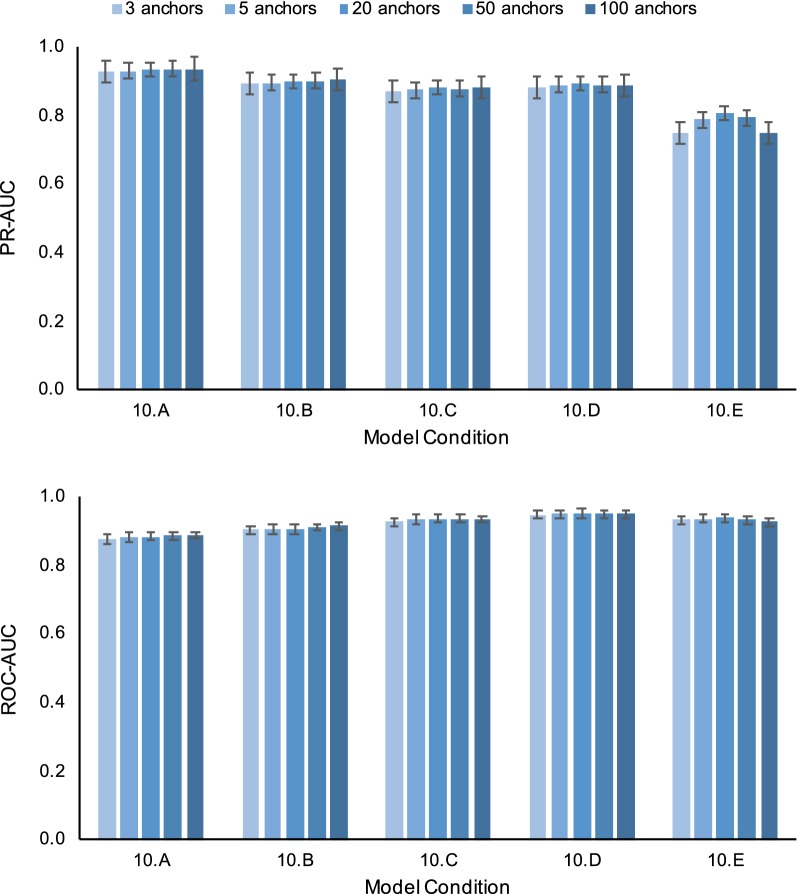
Non-parametric and semi-parametric resampling procedures are widely used to perform support estimation in computational biology and bioinformatics. Among the most widely used methods in this class is the standard bootstrap method, which consists of random sampling with replacement. While not requiring assumptions about any particular parametric model for resampling purposes, the bootstrap and related techniques assume that sites are independent and identically distributed (i.i.d.). The i.i.d. assumption can be an over-simplification for many problems in computational biology and bioinformatics. In particular, sequential dependence within biomolecular sequences is often an essential biological feature due to biochemical function, evolutionary processes such as recombination, and other factors. To relax the simplifying i.i.d. assumption, we propose a new non-parametric/semi-parametric sequential resampling technique that generalizes "Heads-or-Tails" mirrored inputs, a simple but clever technique due to Landan and Graur. The generalized procedure takes the form of random walks along either aligned or unaligned biomolecular sequences. We refer to our new method as the SERES (or "SEquential RESampling") method. To demonstrate the performance of the new technique, we apply SERES to estimate support for the multiple sequence alignment problem. Using simulated and empirical data, we show that SERES-based support estimation yields comparable or typically better performance compared to state-of-the-art methods.
Keywords: Bootstrap; Multiple sequence alignment; Non-parametric; Random walk; Resampling; Semi-parametric; Statistical support.
© The Author(s) 2020.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures

References
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300.
-
- Cannone J, Subramanian S, Schnare M, Collett J, D’Souza L, Du Y, Feng B, Lin N, Madabusi L, Muller K, Pande N, Shang Z, Yu N, Gutell R. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal intron and other RNAs. BMC Bioinform. 2002;3(1):2. doi: 10.1186/1471-2105-3-2. - DOI - PMC - PubMed
-
- Daskalakis C, Roch S. Alignment-free phylogenetic reconstruction. In: Berger B, editor. Research in computational molecular biology. Heidelberg: Springer; 2010. pp. 123–137.
LinkOut - more resources
Full Text Sources
