

Gene Networks with Transcriptional Bursting Recapitulate Rare Transient Coordinated High Expression States in Cancer
- PMID: 32325034
- PMCID: PMC7293108
- DOI: 10.1016/j.cels.2020.03.004
Gene Networks with Transcriptional Bursting Recapitulate Rare Transient Coordinated High Expression States in Cancer
Abstract
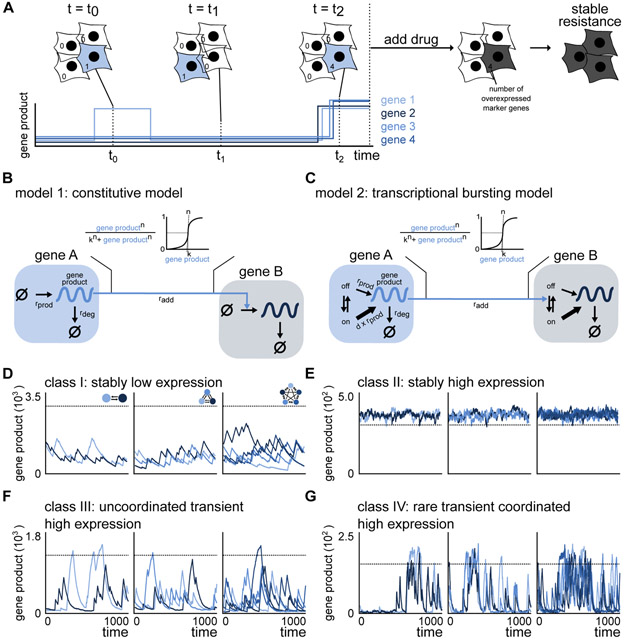
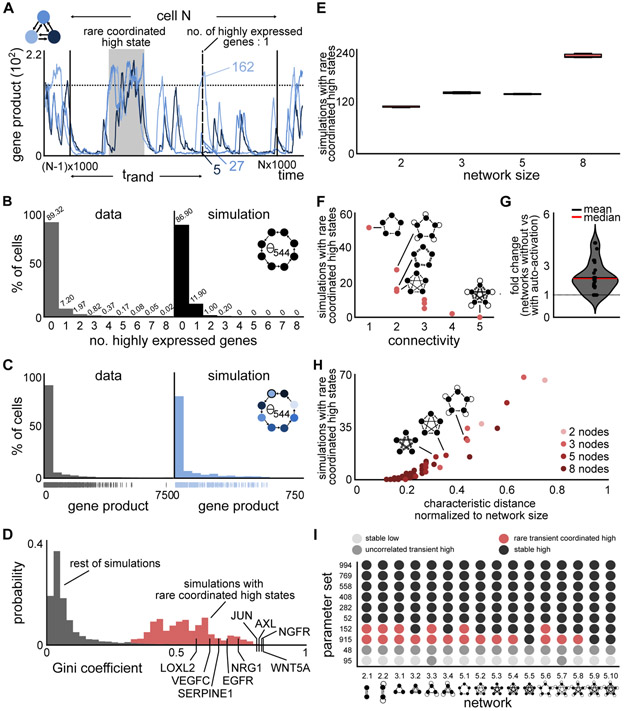
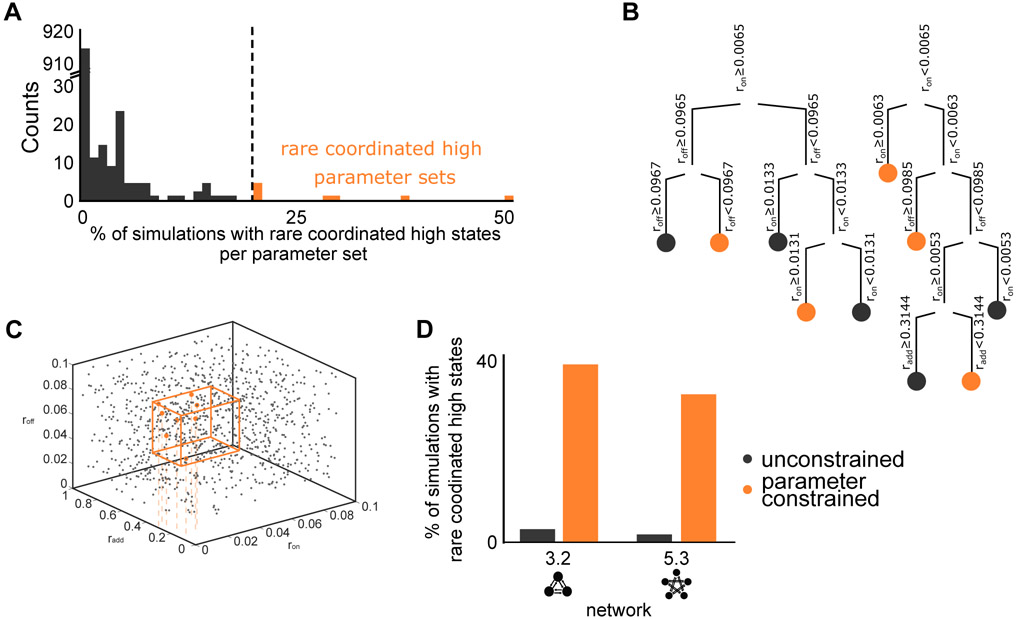
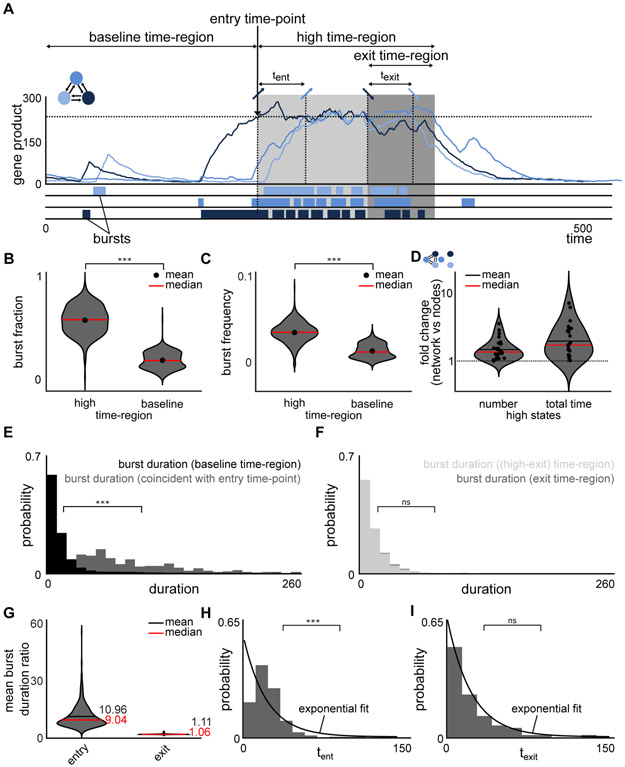
Non-genetic transcriptional variability is a potential mechanism for therapy resistance in melanoma. Specifically, rare subpopulations of cells occupy a transient pre-resistant state characterized by coordinated high expression of several genes and survive therapy. How might these rare states arise and disappear within the population? It is unclear whether the canonical models of probabilistic transcriptional pulsing can explain this behavior, or if it requires special, hitherto unidentified mechanisms. We show that a minimal model of transcriptional bursting and gene interactions can give rise to rare coordinated high expression states. These states occur more frequently in networks with low connectivity and depend on three parameters. While entry into these states is initiated by a long transcriptional burst that also triggers entry of other genes, the exit occurs through independent inactivation of individual genes. Together, we demonstrate that established principles of gene regulation are sufficient to describe this behavior and argue for its more general existence. A record of this paper's transparent peer review process is included in the Supplemental Information.
Keywords: drug resistance; gene expression; melanoma; network; non-genetic; stochasticity.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests A.R. receives royalties related to Stellaris RNA FISH probes. All other authors declare no competing interests.
Figures
References
-
- Breiman L et al. (1984) Classification and Regression Trees (Wadsworth Statistics/Probability). 1 edition. Chapman and Hall/CRC.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
