

AAA+ ATPases in Protein Degradation: Structures, Functions and Mechanisms
- PMID: 32325699
- PMCID: PMC7226402
- DOI: 10.3390/biom10040629
AAA+ ATPases in Protein Degradation: Structures, Functions and Mechanisms
Abstract
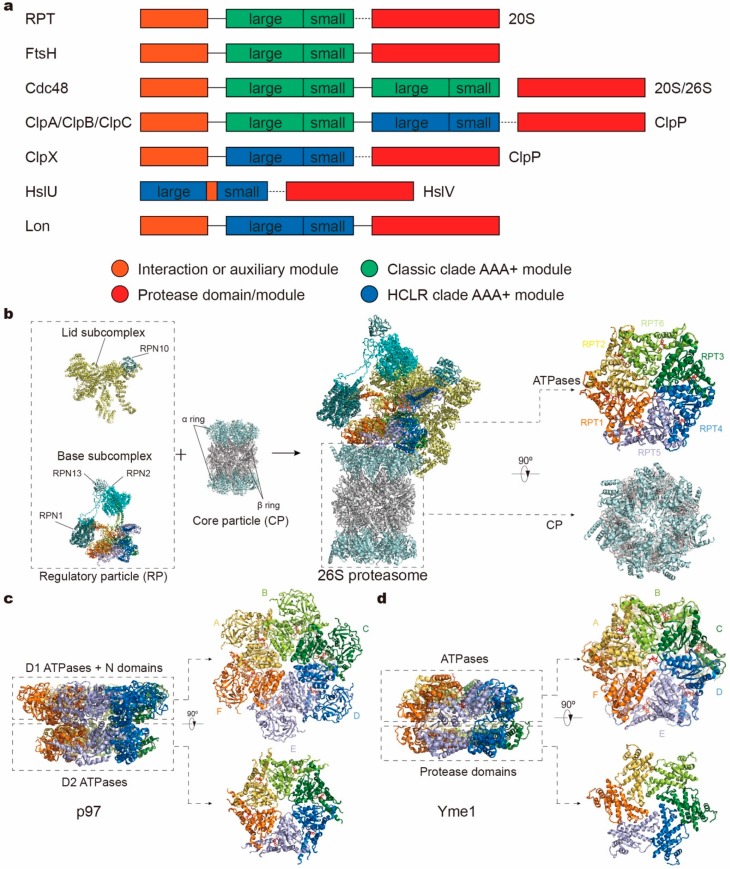
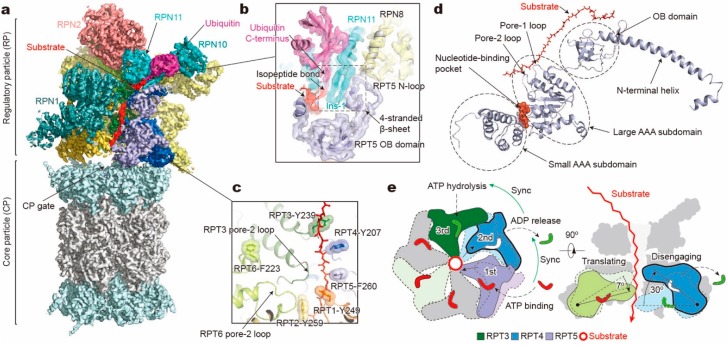
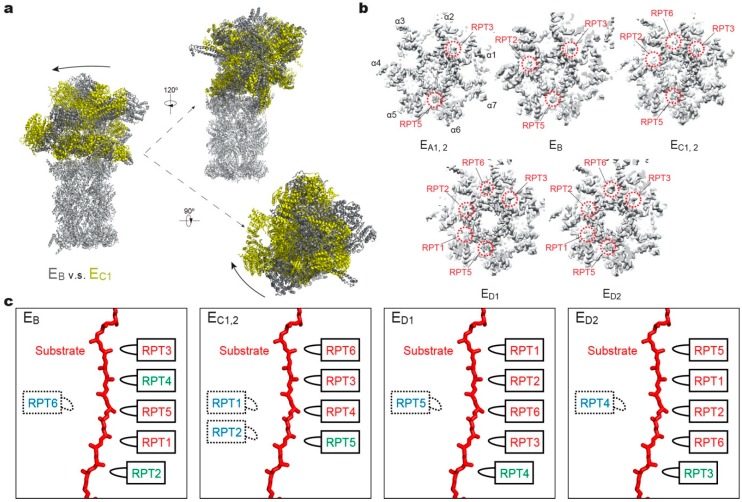
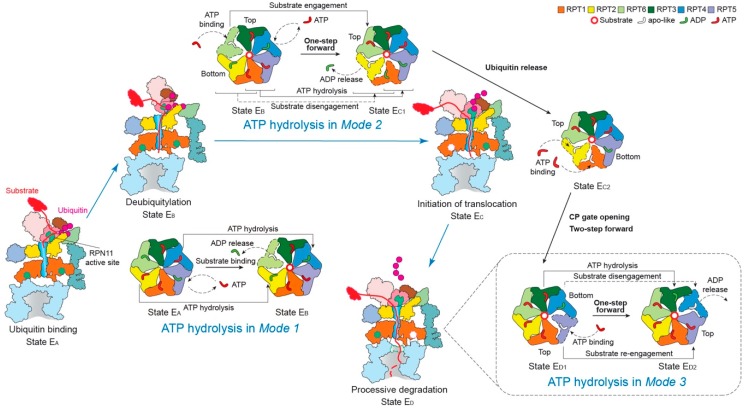
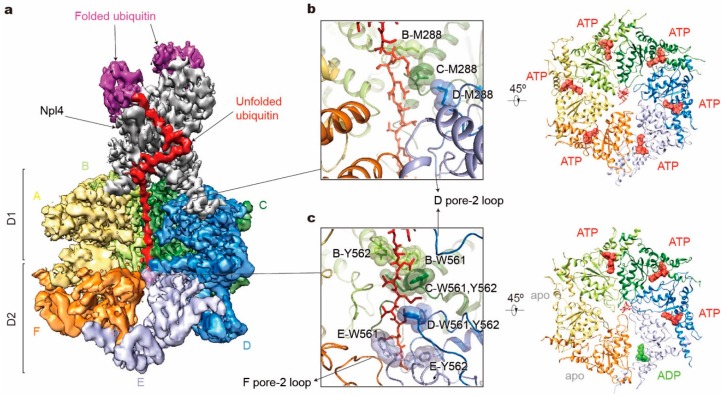
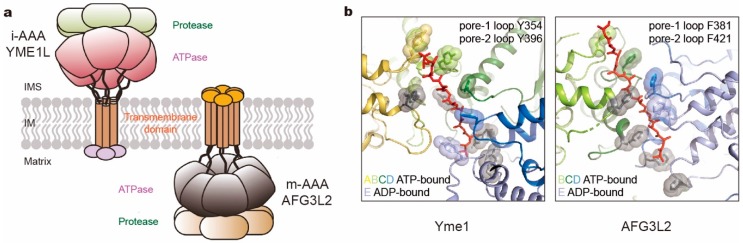
Adenosine triphosphatases (ATPases) associated with a variety of cellular activities (AAA+), the hexameric ring-shaped motor complexes located in all ATP-driven proteolytic machines, are involved in many cellular processes. Powered by cycles of ATP binding and hydrolysis, conformational changes in AAA+ ATPases can generate mechanical work that unfolds a substrate protein inside the central axial channel of ATPase ring for degradation. Three-dimensional visualizations of several AAA+ ATPase complexes in the act of substrate processing for protein degradation have been resolved at the atomic level thanks to recent technical advances in cryogenic electron microscopy (cryo-EM). Here, we summarize the resulting advances in structural and biochemical studies of AAA+ proteases in the process of proteolysis reactions, with an emphasis on cryo-EM structural analyses of the 26S proteasome, Cdc48/p97 and FtsH-like mitochondrial proteases. These studies reveal three highly conserved patterns in the structure-function relationship of AAA+ ATPase hexamers that were observed in the human 26S proteasome, thus suggesting common dynamic models of mechanochemical coupling during force generation and substrate translocation.
Keywords: 26S proteasome; AAA+ ATPase; ATP-dependent proteolysis; Cdc48/p97; mitochondrial protease; substrate translocation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
