

Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration
- PMID: 32326494
- PMCID: PMC7222183
- DOI: 10.3390/antiox9040313
Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration
Abstract
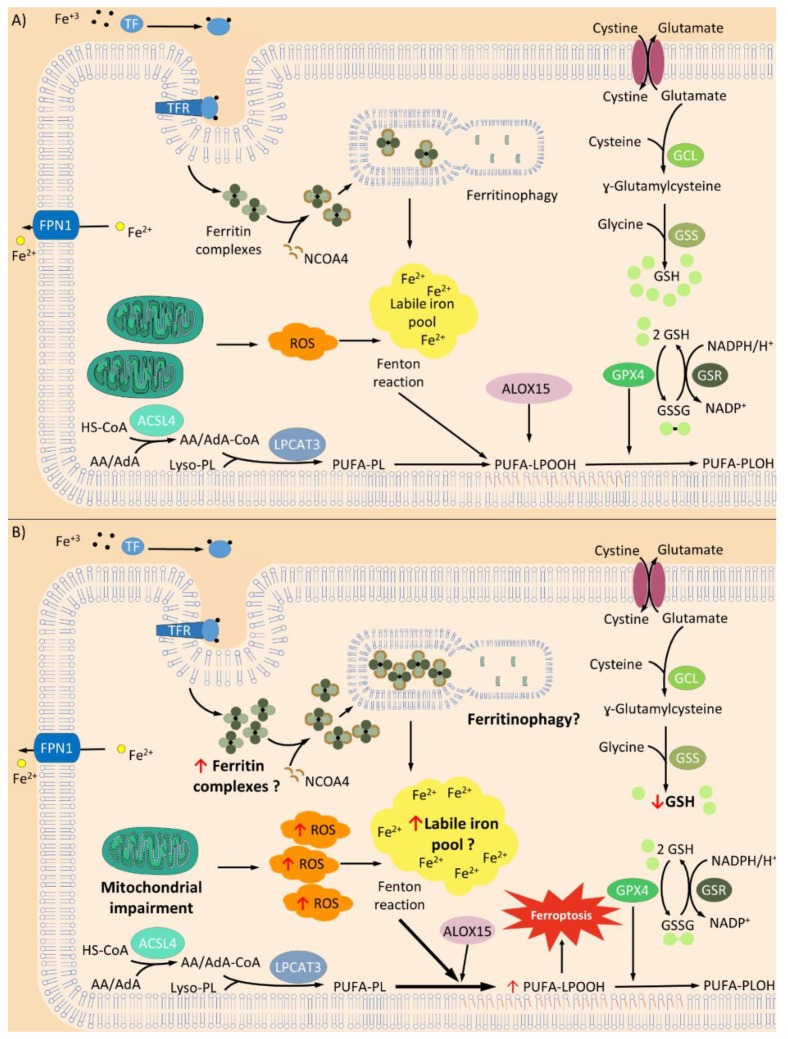
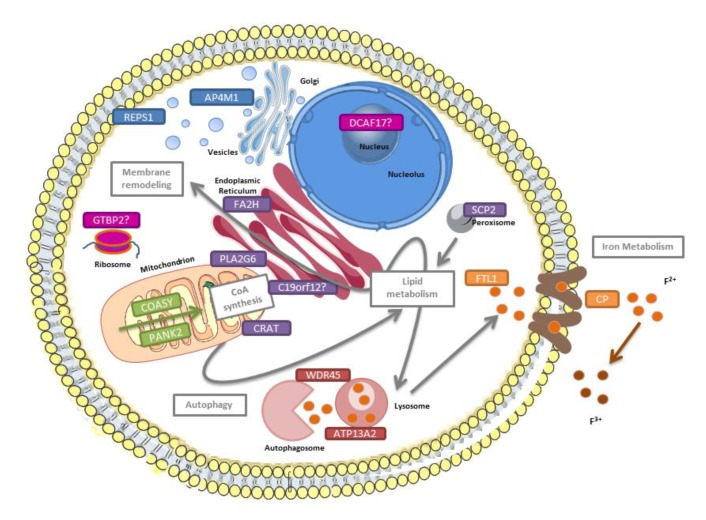
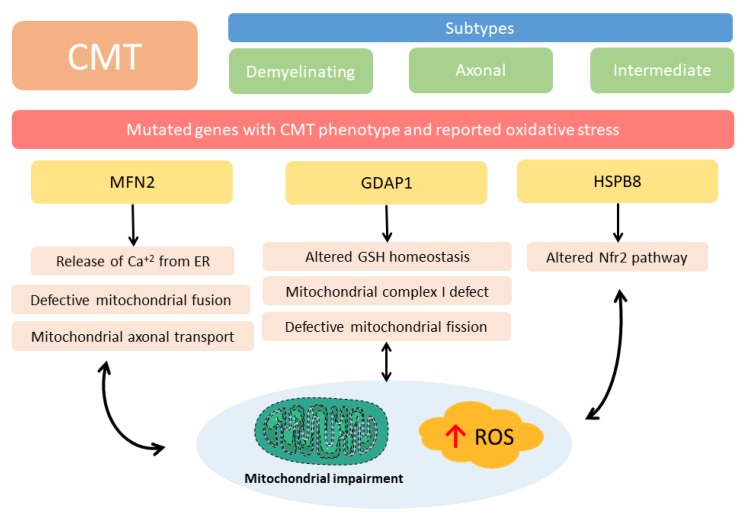
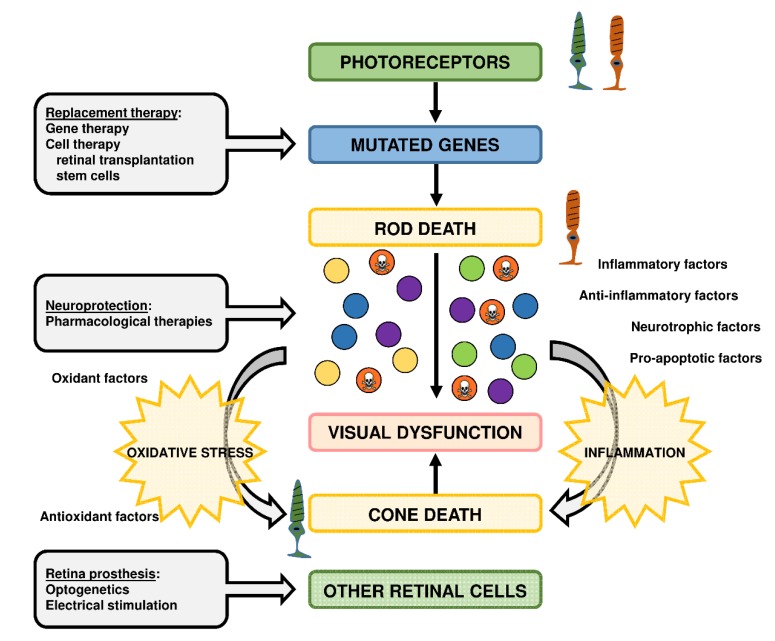
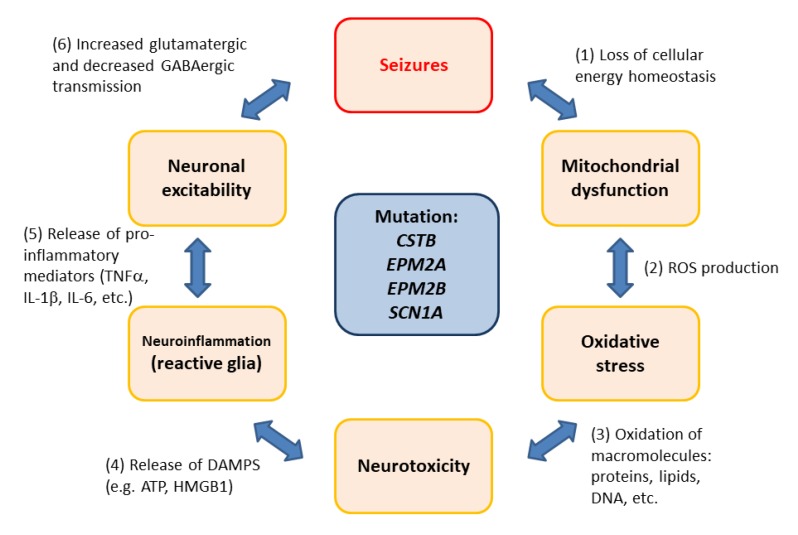
: Oxidative stress is an imbalance between production and accumulation of oxygen reactive species and/or reactive nitrogen species in cells and tissues, and the capacity of detoxifying these products, using enzymatic and non-enzymatic components, such as glutathione. Oxidative stress plays roles in several pathological processes in the nervous system, such as neurotoxicity, neuroinflammation, ischemic stroke, and neurodegeneration. The concepts of oxidative stress and rare diseases were formulated in the eighties, and since then, the link between them has not stopped growing. The present review aims to expand knowledge in the pathological processes associated with oxidative stress underlying some groups of rare diseases: Friedreich's ataxia, diseases with neurodegeneration with brain iron accumulation, Charcot-Marie-Tooth as an example of rare neuromuscular disorders, inherited retinal dystrophies, progressive myoclonus epilepsies, and pediatric drug-resistant epilepsies. Despite the discrimination between cause and effect may not be easy on many occasions, all these conditions are Mendelian rare diseases that share oxidative stress as a common factor, and this may represent a potential target for therapies.
Keywords: Charcot-Marie-Tooth disease (CMT); Dravet syndrome; Friedreich’s ataxia; Lafora disease (LD); Unverricht–Lundborg disease (ULD); inherited retinal dystrophy (IRD); neurodegenerative disorders with brain iron accumulation (NBIA); progressive myoclonus epilepsy (PME).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Cao J.Y., Poddar A., Magtanong L., Lumb J.H., Mileur T.R., Reid M.A., Dovey C.M., Wang J., Locasale J.W., Stone E., et al. A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell Rep. 2019;26:1544–1556. doi: 10.1016/j.celrep.2019.01.043. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
