

Comparing Alternative Single-Step GBLUP Approaches and Training Population Designs for Genomic Evaluation of Crossbred Animals
- PMID: 32328083
- PMCID: PMC7162606
- DOI: 10.3389/fgene.2020.00263
Comparing Alternative Single-Step GBLUP Approaches and Training Population Designs for Genomic Evaluation of Crossbred Animals
Abstract
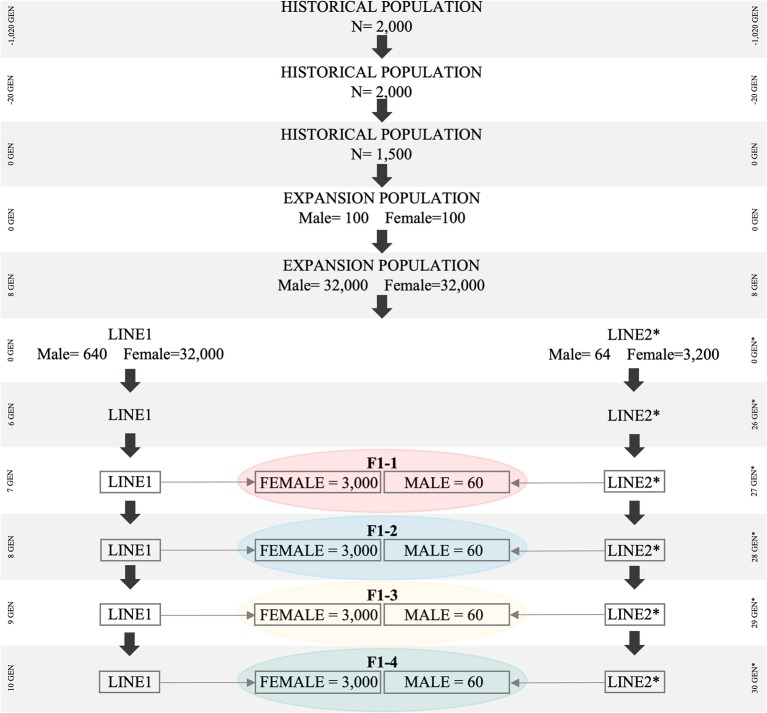
As crossbreeding is extensively used in some livestock species, we aimed to evaluate the performance of single-step GBLUP (ssGBLUP) and weighted ssGBLUP (WssGBLUP) methods to predict Genomic Estimated Breeding Values (GEBVs) of crossbred animals. Different training population scenarios were evaluated: (SC1) ssGBLUP based on a single-trait model considering purebred and crossbred animals in a joint training population; (SC2) ssGBLUP based on a multiple-trait model to enable considering phenotypes recorded in purebred and crossbred training animals as different traits; (SC3) WssGBLUP based on a single-trait model considering purebred and crossbred animals jointly in the training population (both populations were used for SNP weights' estimation); (SC4) WssGBLUP based on a single-trait model considering only purebred animals in the training population (crossbred population only used for SNP weights' estimation); (SC5) WssGBLUP based on a single-trait model and the training population characterized by purebred animals (purebred population used for SNP weights' estimation). A complex trait was simulated assuming alternative genetic architectures. Different scaling factors to blend the inverse of the genomic (G -1) and pedigree ( ) relationship matrices were also tested. The predictive performance of each scenario was evaluated based on the validation accuracy and regression coefficient. The genetic correlations across simulated populations in the different scenarios ranged from moderate to high (0.71-0.99). The scenario mimicking a completely polygenic trait ( 0) yielded the lowest validation accuracy (0.12; for SC3 and SC4). The simulated scenarios assuming 4,500 QTLs affecting the trait and resulted in the greatest GEBV accuracies (0.47; for SC1 and SC2). The regression coefficients ranged from 0.28 (for SC3 assuming polygenic effect) to 1.27 (for SC2 considering 4,500 QTLs). In general, SC3 and SC5 resulted in inflated GEBVs, whereas other scenarios yielded deflated GEBVs. The scaling factors used to combine G -1 and had a small influence on the validation accuracies, but a greater effect on the regression coefficients. Due to the complexity of multiple-trait models and WssGBLUP analyses, and a similar predictive performance across the methods evaluated, SC1 is recommended for genomic evaluation in crossbred populations with similar genetic structures [moderate-to-high (0.71-0.99) genetic correlations between purebred and crossbred populations].
Keywords: WssGBLUP; crossbred performance; simulated dataset; ssGBLUP; training population design.
Copyright © 2020 Alvarenga, Veroneze, Oliveira, Marques, Lopes, Silva and Brito.
Figures







References
-
- Aguilar I., Misztal I., Tsuruta S., Legarra A., Wang H. (2014). PREGSF90 – POSTGSF90: computational tools for the implementation of single-step genomic selection and genome-wide association with ungenotyped individuals in BLUPF90 programs, in Proceedings, 10th World Congress of Genetics Applied to Livestock Production (Vancouver, BC: ).
-
- Bijma P., van Arendonk J. A. M. (1998). Maximizing genetic gain for the sire line of a crossbreeding scheme utilizing both purebred and crossbred information. Anim. Sci. 66, 529–542. 10.1017/S135772980000970X - DOI
-
- Bijma P., Woolliams J. A., Arendonk J. A. M. (2001). Genetic gain of pure line selection and combined crossbred purebred selection with constrained inbreeding. Anim. Sci. 72, 225–232. 10.1017/S1357729800055715 - DOI
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous