

Sample multiplexing for targeted pathway proteomics in aging mice
- PMID: 32332170
- PMCID: PMC7211924
- DOI: 10.1073/pnas.1919410117
Sample multiplexing for targeted pathway proteomics in aging mice
Abstract
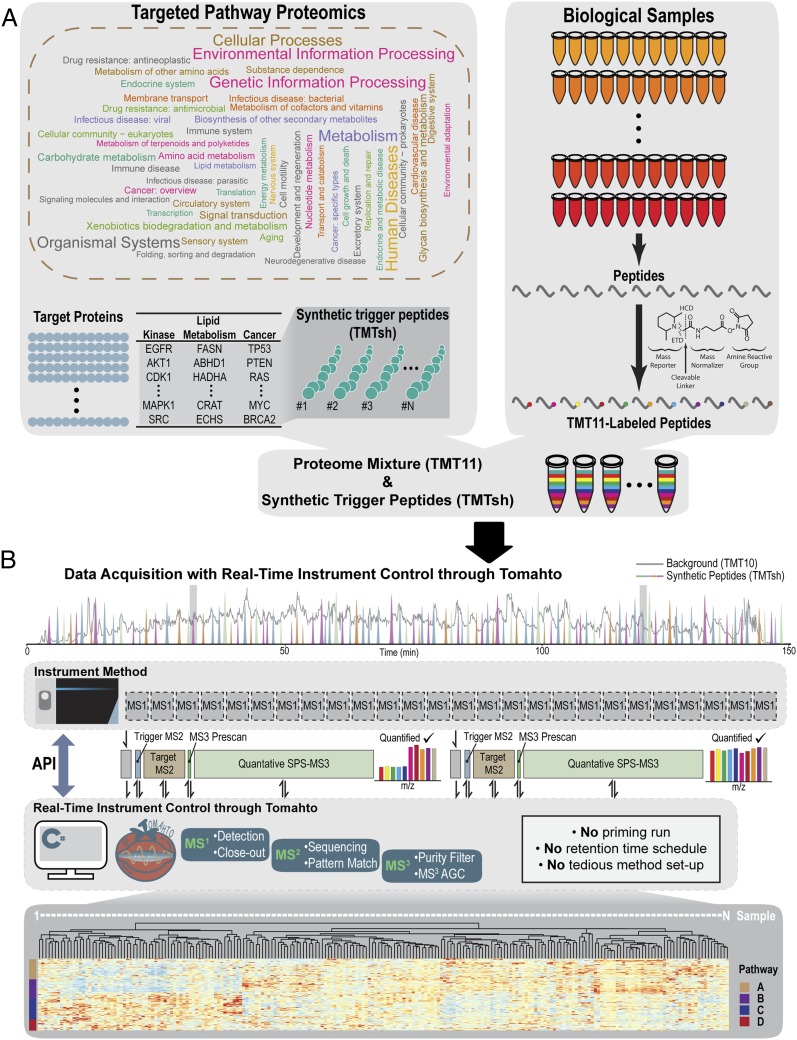
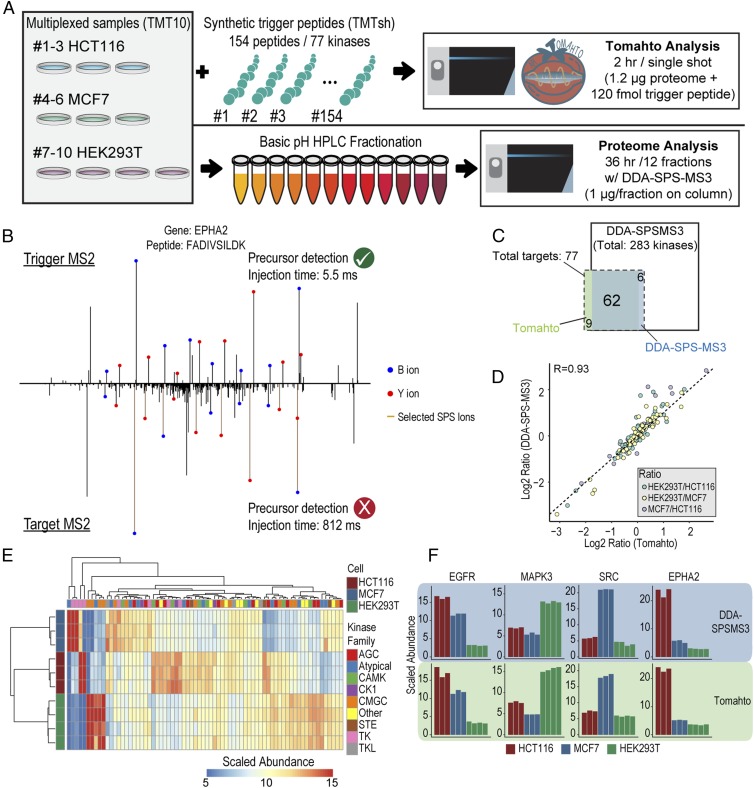
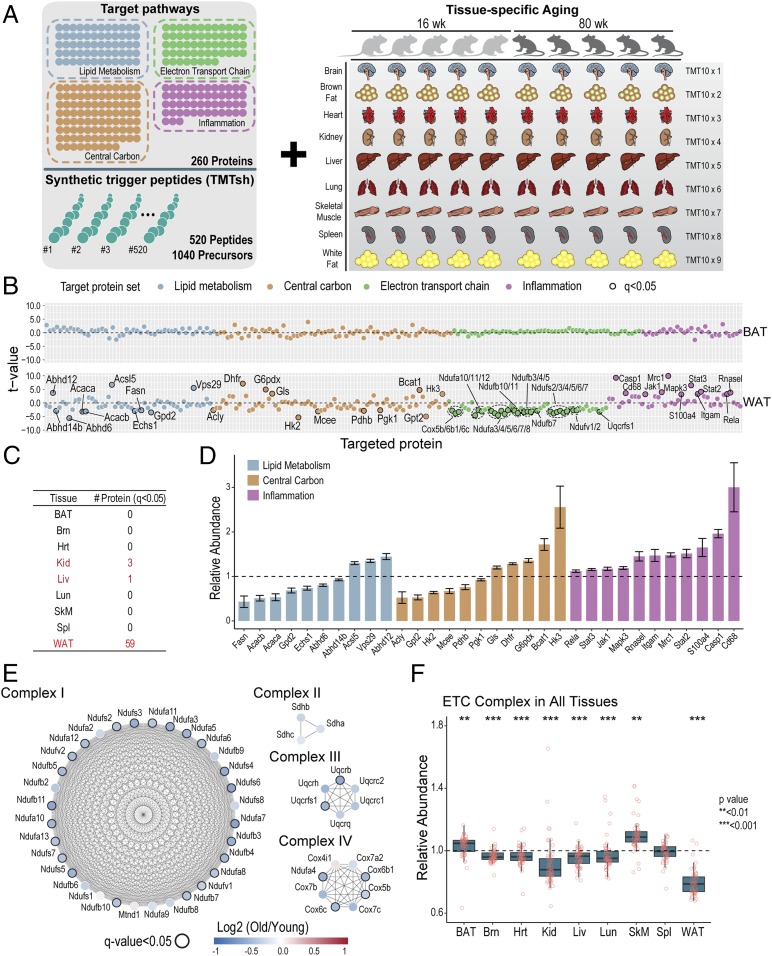
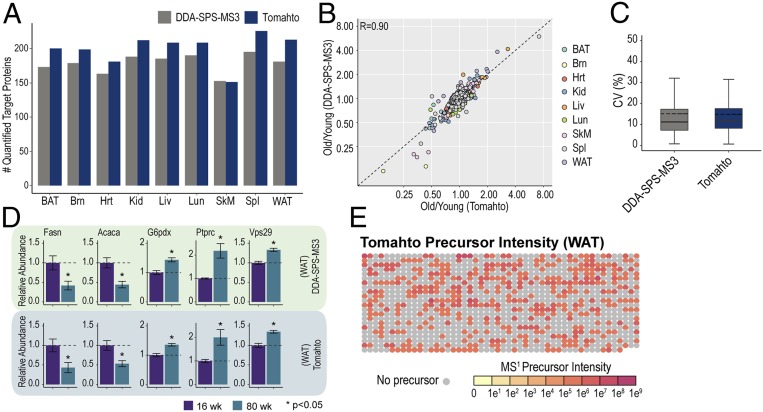
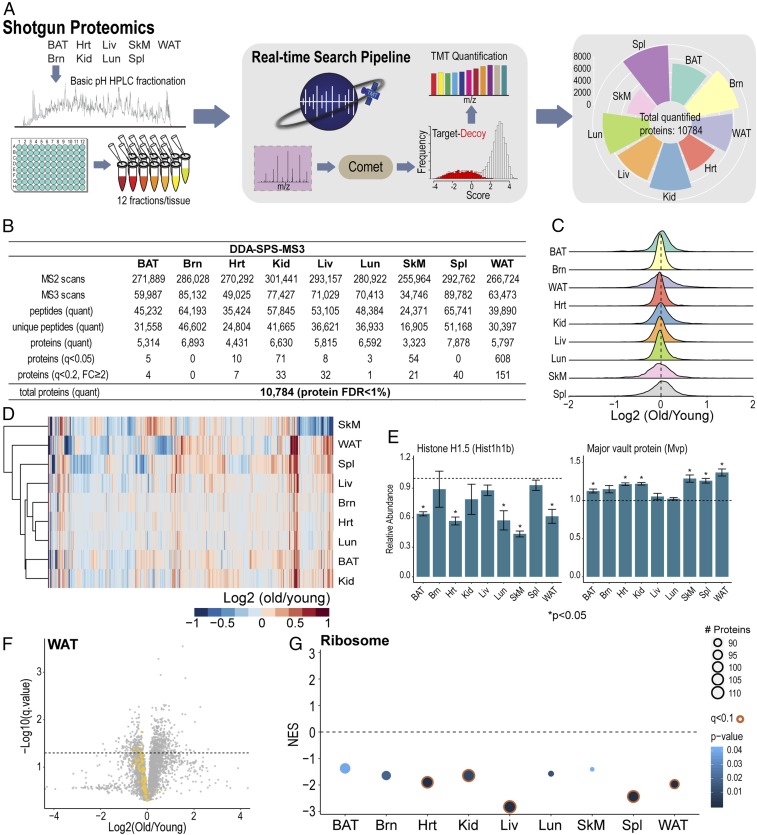
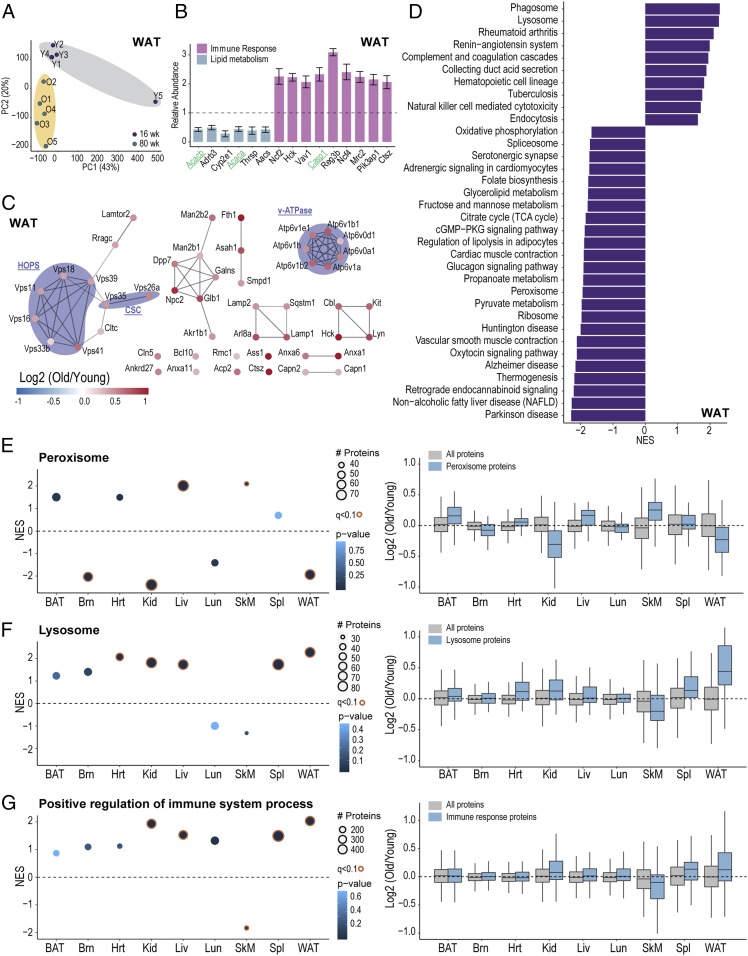
Pathway proteomics strategies measure protein expression changes in specific cellular processes that carry out related functions. Using targeted tandem mass tags-based sample multiplexing, hundreds of proteins can be quantified across 10 or more samples simultaneously. To facilitate these highly complex experiments, we introduce a strategy that provides complete control over targeted sample multiplexing experiments, termed Tomahto, and present its implementation on the Orbitrap Tribrid mass spectrometer platform. Importantly, this software monitors via the external desktop computer to the data stream and inserts optimized MS2 and MS3 scans in real time based on an application programming interface with the mass spectrometer. Hundreds of proteins of interest from diverse biological samples can be targeted and accurately quantified in a sensitive and high-throughput fashion. It achieves sensitivity comparable to, if not better than, deep fractionation and requires minimal total sample input (∼10 µg). As a proof-of-principle experiment, we selected four pathways important in metabolism- and inflammation-related processes (260 proteins/520 peptides) and measured their abundance across 90 samples (nine tissues from five old and five young mice) to explore effects of aging. Tissue-specific aging is presented here and we highlight the role of inflammation- and metabolism-related processes in white adipose tissue. We validated our approach through comparison with a global proteome survey across the tissues, work that we also provide as a general resource for the community.
Keywords: Tomahto; isobaric labeling; real-time instrument control; targeted pathway proteomics; tissue-specific aging.
Copyright © 2020 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no competing interest.
Figures
Comment in
-
Proteomics illuminates fat as key tissue in aging.Proc Natl Acad Sci U S A. 2020 May 12;117(19):10111-10112. doi: 10.1073/pnas.2005988117. Epub 2020 May 4. Proc Natl Acad Sci U S A. 2020. PMID: 32366668 Free PMC article. No abstract available.
References
-
- Gillet L. C., Leitner A., Aebersold R., Mass spectrometry applied to bottom-up proteomics: Entering the high-throughput Era for hypothesis testing. Annu. Rev. Anal. Chem. (Palo Alto, Calif.) 9, 449–472 (2016). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
