

The phylogenetic landscape and nosocomial spread of the multidrug-resistant opportunist Stenotrophomonas maltophilia
- PMID: 32341346
- PMCID: PMC7184733
- DOI: 10.1038/s41467-020-15123-0
The phylogenetic landscape and nosocomial spread of the multidrug-resistant opportunist Stenotrophomonas maltophilia
Abstract
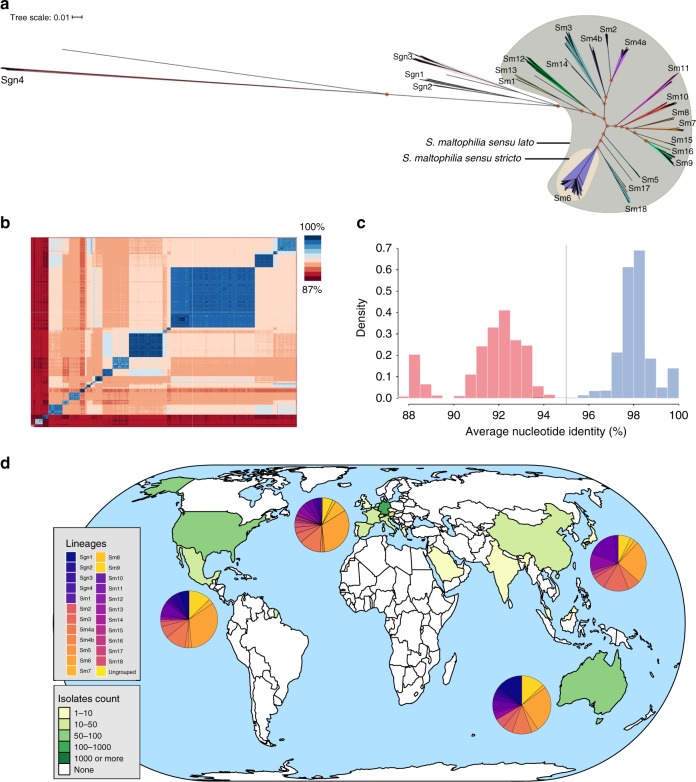
Recent studies portend a rising global spread and adaptation of human- or healthcare-associated pathogens. Here, we analyse an international collection of the emerging, multidrug-resistant, opportunistic pathogen Stenotrophomonas maltophilia from 22 countries to infer population structure and clonality at a global level. We show that the S. maltophilia complex is divided into 23 monophyletic lineages, most of which harbour strains of all degrees of human virulence. Lineage Sm6 comprises the highest rate of human-associated strains, linked to key virulence and resistance genes. Transmission analysis identifies potential outbreak events of genetically closely related strains isolated within days or weeks in the same hospitals.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- World Health Organization. Public health importance of antimicrobial resistance. http://www.who.int/drugresistance/AMR_Importance/en/ (2013). Accessed February 26th, 2020.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
