

Aging-regulated anti-apoptotic long non-coding RNA Sarrah augments recovery from acute myocardial infarction
- PMID: 32341350
- PMCID: PMC7184724
- DOI: 10.1038/s41467-020-15995-2
Aging-regulated anti-apoptotic long non-coding RNA Sarrah augments recovery from acute myocardial infarction
Abstract
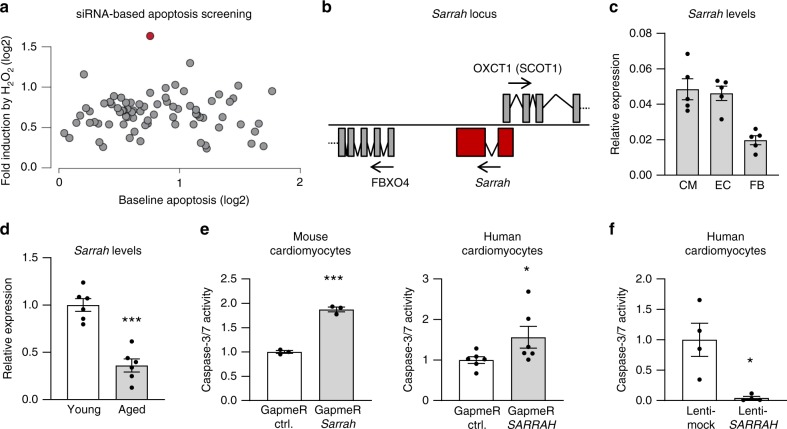
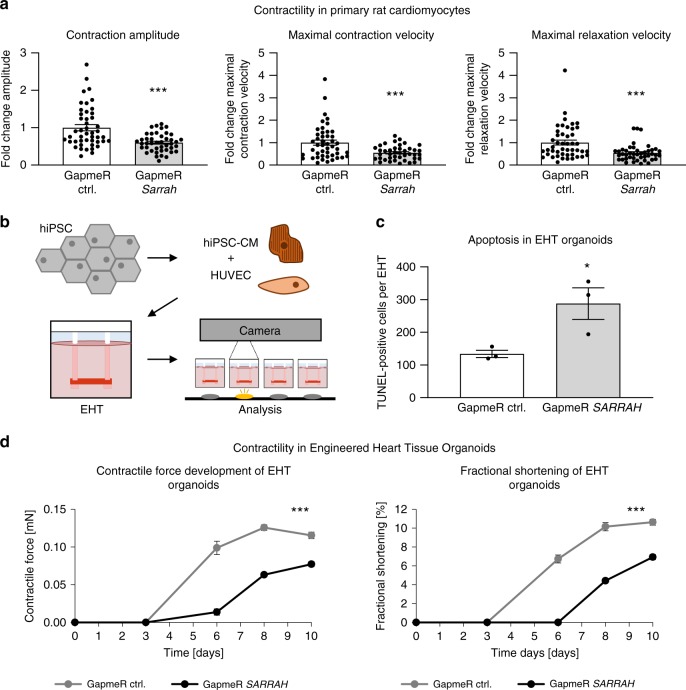
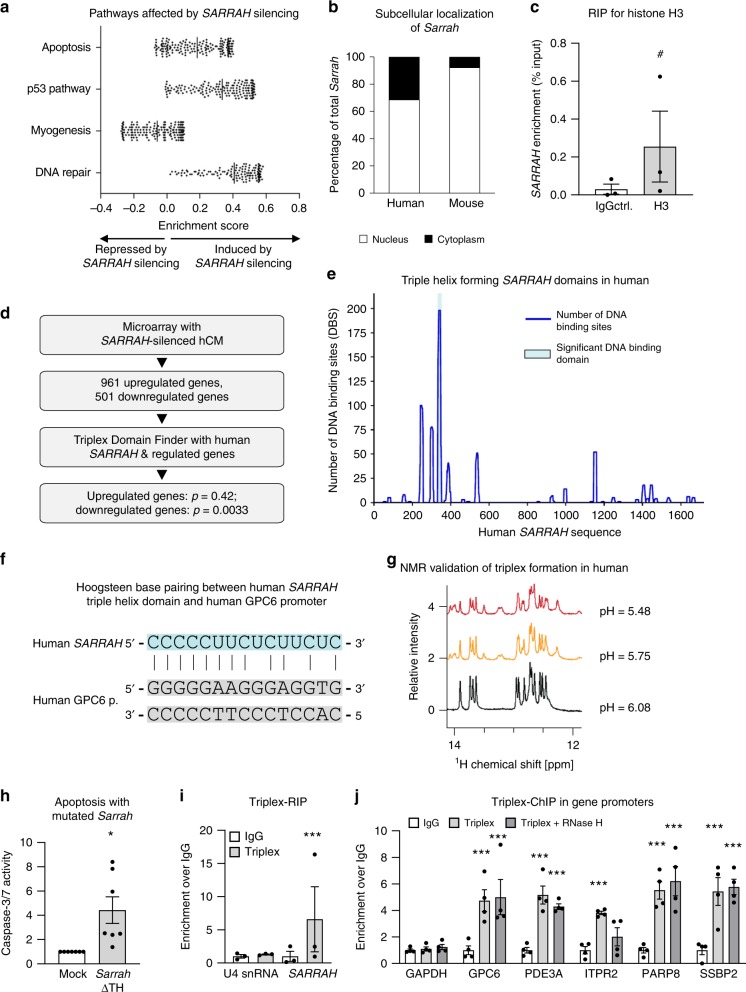
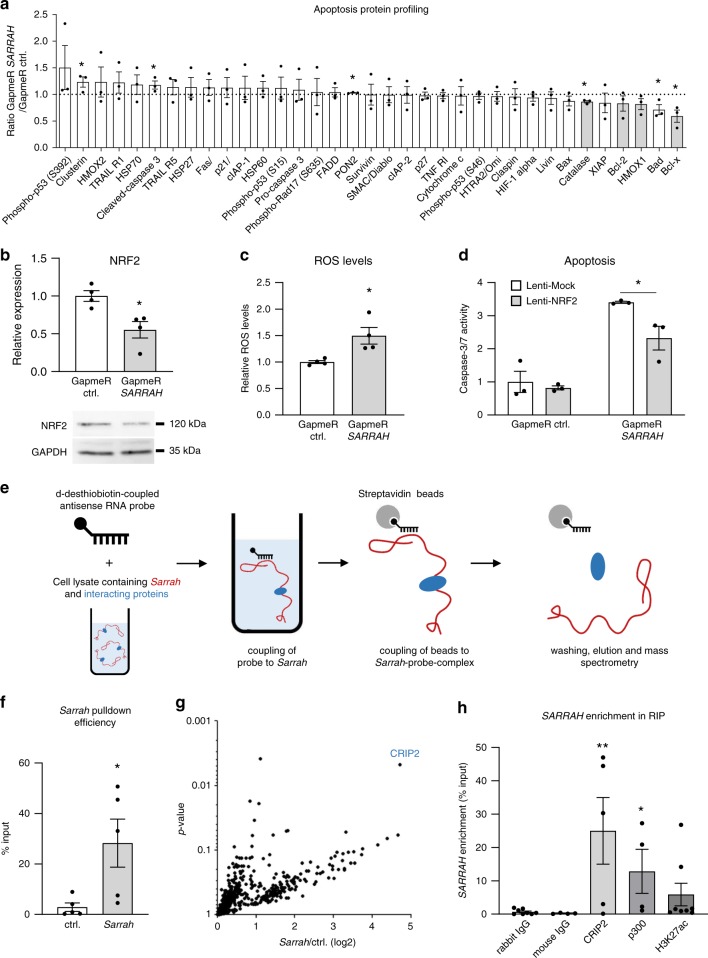
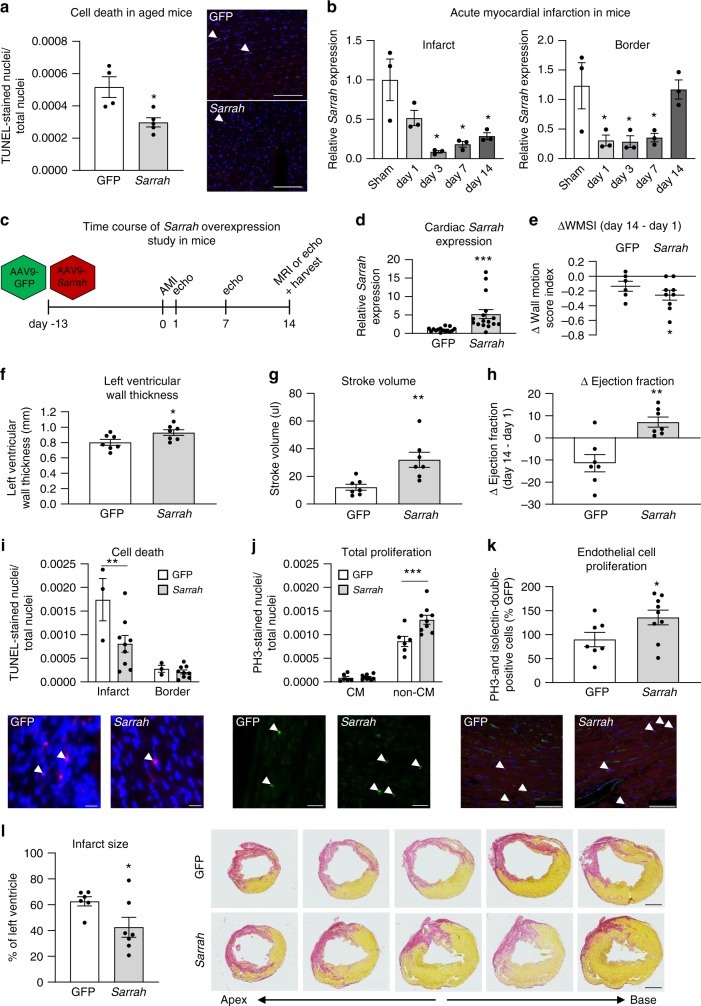
Long non-coding RNAs (lncRNAs) contribute to cardiac (patho)physiology. Aging is the major risk factor for cardiovascular disease with cardiomyocyte apoptosis as one underlying cause. Here, we report the identification of the aging-regulated lncRNA Sarrah (ENSMUST00000140003) that is anti-apoptotic in cardiomyocytes. Importantly, loss of SARRAH (OXCT1-AS1) in human engineered heart tissue results in impaired contractile force development. SARRAH directly binds to the promoters of genes downregulated after SARRAH silencing via RNA-DNA triple helix formation and cardiomyocytes lacking the triple helix forming domain of Sarrah show an increase in apoptosis. One of the direct SARRAH targets is NRF2, and restoration of NRF2 levels after SARRAH silencing partially rescues the reduction in cell viability. Overexpression of Sarrah in mice shows better recovery of cardiac contractile function after AMI compared to control mice. In summary, we identified the anti-apoptotic evolutionary conserved lncRNA Sarrah, which is downregulated by aging, as a regulator of cardiomyocyte survival.
Conflict of interest statement
D.J.T., R.A.B., and S.D. have filed a patent about the therapeutic use of the lncRNA
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
